




A arterite macular linfocítica apresenta‐se mais comumente como máculas hiperpigmentadas nos membros inferiores. A patogênese da doença ainda é incerta e há uma discussão em curso se ela de fato representa uma nova vasculite cutânea ou se trata de uma forma indolente de poliarterite nodosa cutânea.
ObjetivoDescrever achados clínicos, histopatológicos e laboratoriais de pacientes que receberam o diagnóstico de arterite macular linfocítica.
MétodosFoi conduzida uma pesquisa retrospectiva, em que foram revistos os casos seguidos na Clínica de Vasculites da Divisão de Dermatologia, Faculdade de Medicina, Universidade de São Paulo, entre 2005 e 2017. Sete pacientes foram incluídos.
ResultadosTodos os pacientes eram do sexo feminino, entre 9‐46 anos, e tinham máculas hiperpigmentadas principalmente nas pernas. Três pacientes relataram sintomas. As biópsias de pele evidenciavam infiltrados predominantemente linfocíticos que acometiam arteríolas da junção derme‐subcutâneo, bem como um típico anel de fibrina luminal. Nenhum paciente desenvolveu úlceras necróticas, dano neurológico ou manifestações sistêmicas. O seguimento variou de 18 a 151 meses, com duração média de 79 meses.
Limitações do estudoPequena amostra de pacientes e desenho retrospectivo e não controlado.
ConclusõesDe nosso melhor conhecimento, esta série de casos apresenta a maior duração de seguimento já relatada. Durante esse período, nenhum dos pacientes mostrou resolução das lesões nem progressão para vasculite sistêmica. Similaridades entre os achados das biópsias de pele dão apoio à hipótese de que a arterite macular linfocítica é uma forma benigna, incompleta e menos agressiva de poliarterite nodosa cutânea.
A primeira descrição da arterite macular linfocítica (AML) foi feita por Fein et al. em 2003.1 Ela é caracterizada por máculas eritematosas ou hiperpigmentadas, que podem ser redondas, lineares ou mais frequentemente reticuladas, acometem principalmente os membros inferiores e em menor extensão as extremidades superiores e o tronco.2–4 Nódulos não são característicos; entretanto, em alguns casos, indurações subcutâneas sutis têm sido observadas.3,5,6 Livedo racemoso pode aparecer isoladamente ou junto com outros achados dermatológicos.5,7 As lesões seguem um curso indolente e não evoluem para doença sistêmica, embora haja relatos de ulceração cutânea, infartos testiculares e neuropatia.8–11
Ao exame histológico, a AML mostra um denso infiltrado de células mononucleares na parede muscular das arteríolas da junção derme‐subcutâneo, com estreitamento variável de seu lúmen por um típico anel hialinizado de fibrina.12,13 Alguns autores preferem se referir a ela como “arterite trombofílica linfocítica” para enfatizar o suposto mecanismo patogênico associado a essa entidade.7 Na verdade, é questionável se a AML tem características definidoras suficientes para ser considerada uma vasculite distinta da poliarterite nodosa cutânea (PAN‐C), uma arterite necrotizante de vasos de médio calibre com infiltrado predominantemente polimorfonuclear.14
Nosso objetivo foi avaliar retrospectivamente uma série de pacientes com diagnóstico de AML, a fim de analisar suas características clínicas, histopatológicas e laboratoriais.
MétodosO estudo foi aprovado pelo Comitê de Ética da Faculdade de Medicina, Universidade de São Paulo, e todos os participantes preencheram o termo de consentimento informado. Foram incluídos sete pacientes diagnosticados com AML no Ambulatório de Vasculites da Divisão de Dermatologia entre fevereiro de 2005 e setembro de 2017. Os prontuários foram revisados em busca de informações adicionais, como sexo, etnia, idade no início da doença, sintomas associados e história médica prévia, inclusive eventos clínicos trombóticos. No momento do diagnóstico, duas biópsias de pele com profundidade suficiente para incluir o subcutâneo foram feitas em diferentes ocasiões para cada paciente, conforme definido por protocolo institucional. Para este estudo, os aspectos histológicos de cada caso foram revisados por dois dermatopatologistas experientes. A propedêutica laboratorial consistiu de: hemograma completo, funções renal e hepática, velocidade de hemossedimentação, proteína C‐reativa, urinálise, níveis do complemento, eletroforese de proteínas séricas, sorologia para hepatites B e C, fator antinuclear (FAN), fator reumatoide (FR), anticorpo contra DNA dupla hélice, anti‐SSA, anti‐SSB e anticorpos anticitoplasma de neutrófilos. Uma breve pesquisa de trombofilias incluiu: anticoagulante lúpico; anticorpos anticardiolipina (aCL) [imunoglobulina (Ig) M e IgG]; anticorpos anti‐β2 glicoproteína I; proteínas C e S; pesquisa de mutação do fator V de Leiden; mutação do gene da protrombina G20210A; e níveis séricos de antitrombina III, fatores VIII e IX, fibrinogênio, homocisteína e lipoproteína(a) [Lp(a)]. Estudos propedêuticos adicionais de ultrassonografia com Doppler de aorta abdominal, ilíacas e artérias renais foram feitos em três pacientes desta série (pacientes 4, 5 e 6); ultrassonografia com Doppler do sistema venoso, em dois pacientes (pacientes 5 e 6); e eletroneuromiografia, em um paciente (paciente 6).
Para a revisão da literatura, em setembro de 2018 fizemos buscas nas bases de dados PubMed, Web of Science e Scopus com os termos “arterite macular linfocítica”, “arterite macular” ou “arterite trombofílica linfocítica”. Artigos relevantes foram identificados por inspeção manual das listas de referências. Dois investigadores avaliaram títulos e resumos de maneira independente para determinar o potencial de inclusão dos trabalhos. Ao todo, 34 artigos foram analisados. Após excluir um estudo com dados incompletos, foram resumidos os resultados de 23 artigos identificados como relato ou série de casos. Eles serão discutidos posteriormente neste artigo.
ResultadosTodos os pacientes nesta série eram mulheres. Elas foram classificadas de acordo com etnia/cor da pele. Seis eram brancas e uma era negra. A idade variou de 9 a 46 anos, com média ao diagnóstico de 26,71 (DP = 12,44). A duração da doença antes do diagnóstico variou de 6 a 96 meses, com média de 34,42 (DP = 32,88). Hiperpigmentação reticulada persistente foi encontrada em todas as pacientes, seguida por livedo racemoso (duas pacientes) e, em igual frequência, placas eritematosas e indurações subcutâneas sutis (uma paciente cada) (fig. 1). Não houve evidência de ulceração, atrofia branca, nódulos ou púrpura palpável. As pernas foram afetadas em todas as pacientes, enquanto os braços e o tronco em uma paciente cada. As lesões se apresentaram simetricamente quando acometeram as pernas.

Apresentação clínica da arterite macular linfocítica nos membros inferiores. A, Múltiplas máculas lineares e por vezes circulares, veias varicosas e uma cicatriz de biópsia na panturrilha direita; B, Máculas redondas e bem definidas distribuídas nos membros; C, Máculas hipercrômicas bem definidas, algumas das quais levemente infiltradas à palpação e uma cicatriz de biópsia na panturrilha esquerda; D, Máculas eritematosas em padrão reticulado associadas a livedo racemoso.
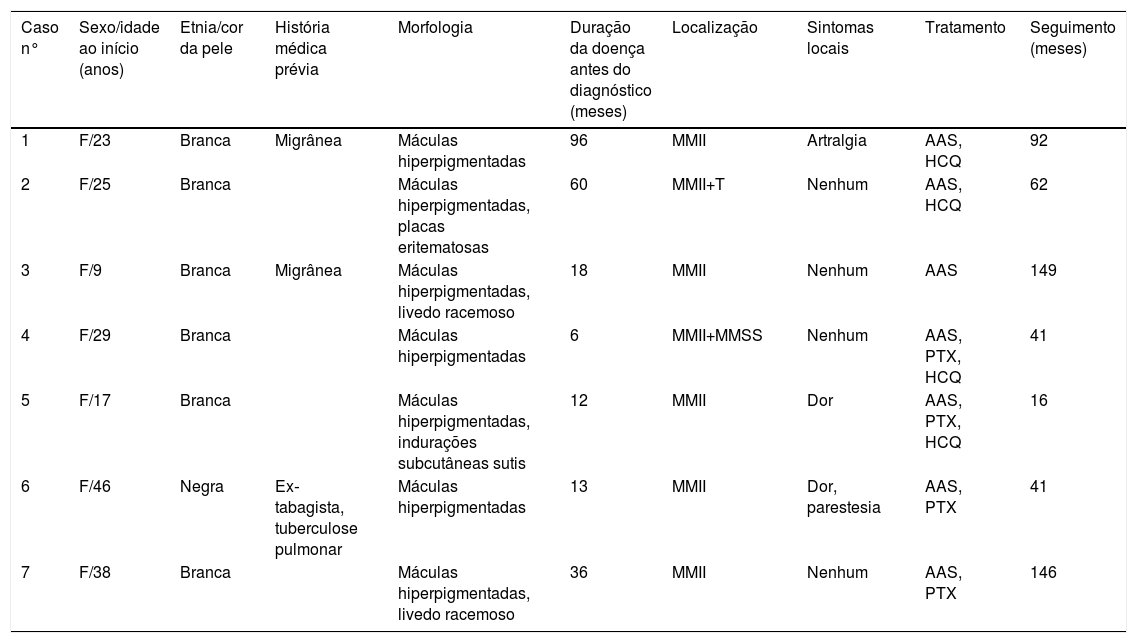
Sintomas referidos no mesmo segmento corporal onde se encontravam as lesões cutâneas foram encontrados em três pacientes. Uma delas (paciente 6) queixou‐se de parestesia e dor; por essa razão, uma eletroneuromiografia foi feita, mas não apresentou alterações, enquanto uma ultrassonografia do sistema venoso dos membros inferiores revelou insuficiência venosa crônica devido a um refluxo primário da safena magna na perna esquerda. Estudos de ultrassonografia com Doppler da aorta abdominal, artérias ilíacas e renais das pacientes 4, 5 e 6 resultaram normais, assim como uma ultrassonografia do sistema venoso dos membros inferiores da paciente 5. Nenhuma das pacientes tinha história de trombose arterial ou venosa. Uma das pacientes era ex‐tabagista, diagnosticada e tratada para tuberculose pulmonar quatro anos antes da admissão. Esfregaço para baciloscopia do escarro e tomografia computadorizada do tórax não mostraram sinais de doença ativa. Duas pacientes foram diagnosticadas com migrânea e foram acompanhadas pela equipe de neurologia do hospital. Dados demográficos, achados histopatológicos, tratamento e duração do seguimento estão representados na tabela 1.
Dados epidemiológicos e características clínicas de sete pacientes diagnosticados com arterite macular linfocítica
| Caso n° | Sexo/idade ao início (anos) | Etnia/cor da pele | História médica prévia | Morfologia | Duração da doença antes do diagnóstico (meses) | Localização | Sintomas locais | Tratamento | Seguimento (meses) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F/23 | Branca | Migrânea | Máculas hiperpigmentadas | 96 | MMII | Artralgia | AAS, HCQ | 92 |
| 2 | F/25 | Branca | Máculas hiperpigmentadas, placas eritematosas | 60 | MMII+T | Nenhum | AAS, HCQ | 62 | |
| 3 | F/9 | Branca | Migrânea | Máculas hiperpigmentadas, livedo racemoso | 18 | MMII | Nenhum | AAS | 149 |
| 4 | F/29 | Branca | Máculas hiperpigmentadas | 6 | MMII+MMSS | Nenhum | AAS, PTX, HCQ | 41 | |
| 5 | F/17 | Branca | Máculas hiperpigmentadas, indurações subcutâneas sutis | 12 | MMII | Dor | AAS, PTX, HCQ | 16 | |
| 6 | F/46 | Negra | Ex‐tabagista, tuberculose pulmonar | Máculas hiperpigmentadas | 13 | MMII | Dor, parestesia | AAS, PTX | 41 |
| 7 | F/38 | Branca | Máculas hiperpigmentadas, livedo racemoso | 36 | MMII | Nenhum | AAS, PTX | 146 |
AAS, ácido acetilsalicílico; F, feminino; HCQ, hidroxicloroquina; MMII, membros inferiores; MMSS, membros superiores; PTX, pentoxifilina; T, tronco.
A avaliação laboratorial mostrou poucas alterações, como baixa atividade de proteína S 28% (VR 55%‐160%) (paciente 3); FAN de 1:80 com padrão nuclear pontilhado fino e nível de fibrinogênio ligeiramente elevado 406mg/dL (VR 150‐400mg/dL) (paciente 5). Duas pacientes apresentaram títulos significativamente positivos de anticorpos aCL IgM em testes repetidos (pacientes 1 e 3), uma das quais apresentava também títulos positivos de FR 41,7 UI/mL (VR < 20 UI/mL) (paciente 1); A Lp(a) encontrava‐se elevada em três pacientes 107, 75 e 50mg/dL (VR < 30mg/dL) (pacientes 2, 4 e 6, respectivamente). O exame histológico das biópsias de pele revelou achados similares em todos os espécimes: epiderme sem alterações dignas de nota; infiltrado linfocítico ao redor de pequenas artérias na derme reticular profunda e subcutâneo superficial e um anel hialinizado de fibrina na luz dos vasos em todos os sete casos, além de oclusão do lúmen vascular por trombo de fibrina em duas pacientes (fig. 2). A composição muscular dos vasos de médio calibre foi mais bem visualizada com a coloração de resorcina‐fucsina, que revelou uma lâmina elástica interna focalmente descontínua e interrompida em uma paciente. Neutrófilos não estavam presentes. Imunofluorescência direta foi feita em três pacientes e apenas em uma revelou depósitos de C3 na parede de vasos dérmicos. Estudo de imuno‐histoquímica com anticorpos anti‐BCG foi conduzido em três pacientes e estava normal.
; B, Estreitamento da luz do vaso por espessamento subintimal (Hematoxilina & eosina, 100×); C, Presença de inflamação densa que envolve as paredes de uma arteríola no subcutâneo superficial (Hematoxilina & eosina, 200×); D, Vaso hipodérmico com trombo organizado (Hematoxilina & eosina, 200x).")
Exame histológico de biópsia de lesões compatíveis com arterite macular linfocítica nas pernas. A, Uma artéria cutânea de pequeno calibre na derme profunda circundada e infiltrada por linfócitos (Hematoxilina & eosina, 200×); B, Estreitamento da luz do vaso por espessamento subintimal (Hematoxilina & eosina, 100×); C, Presença de inflamação densa que envolve as paredes de uma arteríola no subcutâneo superficial (Hematoxilina & eosina, 200×); D, Vaso hipodérmico com trombo organizado (Hematoxilina & eosina, 200x).
O tratamento medicamentoso usado isolado ou em combinação incluiu ácido acetilsalicílico em todas as sete pacientes, além de pentoxifilina e hidroxicloroquina em uma paciente. O seguimento variou de 18‐151 meses, com duração média de 79 meses. Durante esse período, nenhuma paciente mostrou resolução das lesões, nem progrediu para vasculite sistêmica.
DiscussãoOs resultados do presente estudo estão de acordo com a literatura revisada. A principal apresentação clínica da AML ocorreu sob a forma de máculas hiperpigmentadas crônicas e persistentes, embora outros achados dermatológicos, como livedo racemoso, placas eritematosas e indurações subcutâneas sutis, tenham sido detectados. Houve predileção pelas extremidades inferiores. No entanto, a frequência de sintomas reportados foi maior nesta série do que em relatos prévios. As características clínicas dos pacientes com AML já publicados na literatura, bem como seus achados laboratoriais, estão resumidos no material suplementar (Apêndice A).
Houve relatos isolados de casos associando AML à infecção por vírus do HIV ou hepatite B, artrite reumatoide e lúpus eritematoso discoide e a medicações como minociclina e tabaco.6,12,15–18 Várias alterações laboratoriais têm sido descritas, mas pela primeira vez descrevemos níveis elevados de Lp(a) em três pacientes diagnosticados com AML. A Lp(a) está envolvida na modulação da agregação plaquetária, no recrutamento de células inflamatórias e na indução do remodelamento vascular.19 De modo similar, nosso grupo de estudos descobriu níveis elevados de Lp(a) em pacientes com vasculopatia livedoide, uma vasculopatia trombo‐oclusiva de vasos dérmicos que usualmente se apresenta com púrpuras reticuladas recorrentes nas pernas e nos pés, as quais, por sua vez, levam a lesões ulceradas e cicatrizes estelares atróficas e de cor marfim, conhecidas como atrofia branca.20
Duas pacientes nesta série tinham história de migrânea com aura. Em casos semelhantes, é essencial excluir a síndrome de Sneddon, uma vasculopatia trombótica não inflamatória caracterizada pela associação de livedo, que acomete tronco e/ou glúteos, e eventos isquêmicos cerebrovasculares, já que metade desses pacientes apresenta tal padrão de cefaleia.21 Ambas apresentavam duas biópsias de pele cada em diferentes ocasiões e seguimento superior a cinco anos. Uma delas apresentou também títulos positivos de anticorpos aCL de classe IgM em testes repetidos com pelo menos 12 semanas de intervalo. Embora não tivesse história de trombocitopenia, eventos trombóticos no passado ou manifestações obstétricas, procuramos descartar síndrome antifosfolípide. Classicamente, os achados histológicos nessa síndrome são de trombose sem evidência significativa de inflamação na parede vascular.22
Achados histológicos típicos foram vistos em todas as biópsias de pele feitas neste grupo (fig. 3), inclusive estudos de imunofluorescência direta negativos para depósitos de IgM, IgG e IgA na membrana basal e em torno dos vasos. Como relatado previamente e de maneira contrária à descrição original, em alguns casos a coloração para tecidos elásticos revelou uma disrupção na lâmina elástica interna.15,23,24 Um dos diagnósticos diferenciais histológicos da AML é a doença de Buerger (tromboangeíte obliterante), uma doença inflamatória segmentar não aterosclerótica que frequentemente afeta artérias de pequeno e médio calibre, veias e nervos nas extremidades superiores e inferiores. História atual ou prévia de tabagismo é imprescindível para o diagnóstico, uma vez que se trata do principal agente associado à doença.25,26 Apesar de uma de nossas pacientes apresentar antecedente de uso de tabaco, desfavorece esse diagnóstico a ausência de mudanças na coloração da pele e na temperatura dos membros acometidos, ulcerações isquêmicas e gangrena.
.")
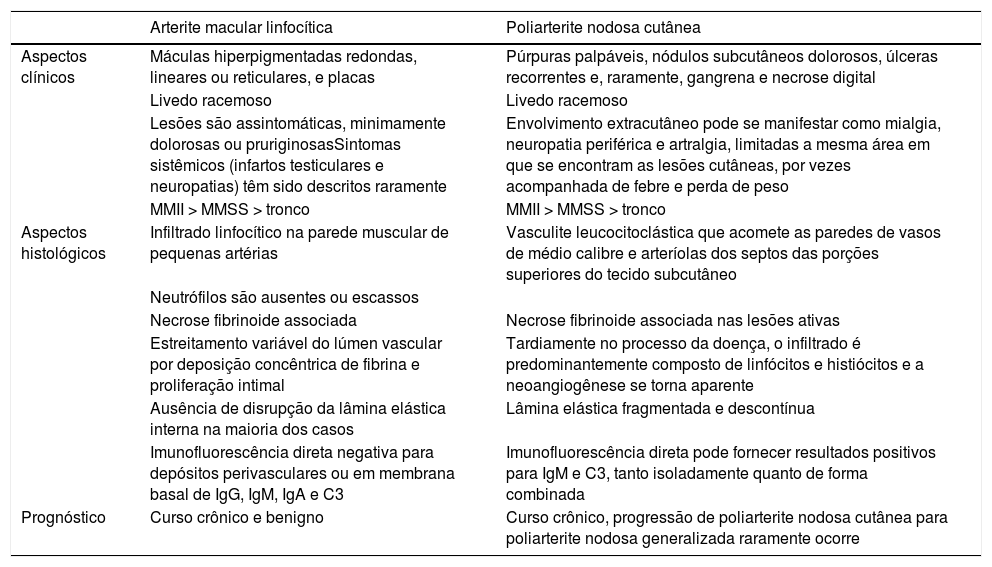
Enquanto isso, lesões agudas de PAN‐C, a variante da poliarterite nodosa limitada à pele, exibem vasculite necrotizante fibrinoide de artérias de pequeno e médio calibre localizadas na junção derme‐subcutâneo com um infiltrado predominantemente composto de células polimorfonucleares. Clinicamente, PAN‐C se apresenta com maior frequência como nódulos subcutâneos dolorosos e úlceras nos membros inferiores, geralmente em associação com livedo racemoso. Sintomas extracutâneos, como neuropatia periférica, mialgia e artralgia, são limitados à mesma área em que estão situadas as lesões cutâneas.27–29 Características relevantes de ambas AML e PAN‐C estão apresentadas na tabela 2.
Aspectos clínicos e histológicos da arterite macular linfocítica e poliarterite nodosa cutânea
| Arterite macular linfocítica | Poliarterite nodosa cutânea | |
|---|---|---|
| Aspectos clínicos | Máculas hiperpigmentadas redondas, lineares ou reticulares, e placas | Púrpuras palpáveis, nódulos subcutâneos dolorosos, úlceras recorrentes e, raramente, gangrena e necrose digital |
| Livedo racemoso | Livedo racemoso | |
| Lesões são assintomáticas, minimamente dolorosas ou pruriginosasSintomas sistêmicos (infartos testiculares e neuropatias) têm sido descritos raramente | Envolvimento extracutâneo pode se manifestar como mialgia, neuropatia periférica e artralgia, limitadas a mesma área em que se encontram as lesões cutâneas, por vezes acompanhada de febre e perda de peso | |
| MMII > MMSS > tronco | MMII > MMSS > tronco | |
| Aspectos histológicos | Infiltrado linfocítico na parede muscular de pequenas artérias | Vasculite leucocitoclástica que acomete as paredes de vasos de médio calibre e arteríolas dos septos das porções superiores do tecido subcutâneo |
| Neutrófilos são ausentes ou escassos | ||
| Necrose fibrinoide associada | Necrose fibrinoide associada nas lesões ativas | |
| Estreitamento variável do lúmen vascular por deposição concêntrica de fibrina e proliferação intimal | Tardiamente no processo da doença, o infiltrado é predominantemente composto de linfócitos e histiócitos e a neoangiogênese se torna aparente | |
| Ausência de disrupção da lâmina elástica interna na maioria dos casos | Lâmina elástica fragmentada e descontínua | |
| Imunofluorescência direta negativa para depósitos perivasculares ou em membrana basal de IgG, IgM, IgA e C3 | Imunofluorescência direta pode fornecer resultados positivos para IgM e C3, tanto isoladamente quanto de forma combinada | |
| Prognóstico | Curso crônico e benigno | Curso crônico, progressão de poliarterite nodosa cutânea para poliarterite nodosa generalizada raramente ocorre |
MMII, membros inferiores; MMSS, membros superiores.
Diversos tratamentos para AML têm sido relatados com resultados variáveis. Esteroides tópicos já foram tentados sem melhoria clínica.15,24,30 Além disso, não houve resposta a baixas doses de aspirina, clopidogrel e varfarina. De fato, em dois casos as lesões progrediram mesmo durante o uso da medicação. Até 80% de melhoria no aspecto das lesões foi demostrado durante o uso de dapsona 100mg/dia e resolução total após o uso de prednisona em baixas doses, metotrexato, bem como de colchicina, embora a duração do seguimento relatada nesse último caso tenha sido de apenas oito meses.30–32 Os autores escolheram ácido acetilsalicílico e pentoxifilina como tratamento de primeira linha para a AML. Em quatro pacientes, a hidroxicloroquina foi acrescentada por persistência dos sintomas ou por extensão das lesões para segmentos além das pernas e elas se mantiveram estáveis. Não se observou regressão das lesões cutâneas maculares, nem surgimento de úlceras necróticas, dano neurológico ou manifestações sistêmicas. Do conhecimento dos autores, a duração do seguimento nesta série é a maior já relatada na literatura.
ConclusõesEste estudo descreve sete pacientes cuja apresentação clínica e histopatológica é compatível com AML. Dada a sobreposição de aspectos clínicos, existe um debate em curso se a AML poderia ou não ser considerada uma forma precursora ou indolente de PAN‐C. Levando em consideração a existência de uma já bem estudada transição de um infiltrado predominantemente neutrofílico para um infiltrado linfocítico nos estados subagudos e reparativos de PAN‐C, assim como o fato de que alguns pacientes com AML podem exibir características histológicas intermediárias, como necrose fibrinoide e disrupção da lâmina elástica interna, os autores acreditam ser razoável considerá‐las como pertencentes a um mesmo espectro de doença. Propõe‐se que a AML possa representar uma resposta imune menos agressiva ao componente arterial dos vasos de médio calibre, a qual é insuficiente para provocar ulcerações, cicatrizes ou sintomas inconvenientes na maioria das vezes. Ao longo do tempo, mais pesquisas sobre sua etiopatogênese podem elucidar se de fato existe alguma relação entre ambas.
Suporte financeiroNenhum.
Contribuição dos autoresThamara Cristiane Alves Batista Morita: Análise estatística; aprovação da versão final do manuscrito; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Gabriela Franco Sturzeneker Trés: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Paulo Ricardo Criado: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Como citar este artigo: Morita TCAB, Trés GFS, Criado PR. Is macular lymphocytic arteritis limited to the skin? Long‐term follow‐up of seven patients. An Bras Dermatol. 2020;95:32–9.
Trabalho realizado no Departamento de Dermatologia, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil.
