


A síndrome de Coffin‐Siris é doença congênita clínica e geneticamente heterogênea, marcada por características faciais grosseiras, deficiência intelectual, hipoplasia do quinto quirodáctilo e aplasia ou hipoplasia ungueal.
Paciente do sexo masculino, de 7 meses de vida, foi atendido no Serviço de Dermatologia em virtude de doença ungueal congênita. A criança havia sido diagnosticada com megacisterna magna, forame oval permeável, hipoplasia renal direita e desenvolvimento psicomotor ligeiramente atrasado com risco de comprometimento do desenvolvimento cognitivo. O exame físico revelou displasia de todas as unhas e anoníquia ou microníquia do 3°, 4° e 5° pododáctilos e do 4° e 5° quirodáctilos (fig. 1 A‐B). O paciente apresentava traços faciais característicos, com ponte nasal ampla, boca larga e lábios grossos.
 e radiográficas (C‐D) mostrando hipoplasia das falanges distais do 2°, 3°, 4° e 5° quirodáctilos, ausência de falanges distais no 2°, 3°, 4° e 5° pododáctilos e hipoplasia da falange distal do 1° pododáctilo.")
A radiografia das mãos e dos pés revelou hipoplasia das falanges distais do 2°, 3°, 4° e 5° dedos das mãos, ausência de falanges distais nos 2°, 3°, 4° e 5° dedos dos pés e hipoplasia da falange distal do 1° dedo dos pés (fig. 1 C‐D).
Estudo genético foi realizado, com suspeita de síndrome de Coffin‐Siris, e revelou mutação heterozigótica de novo em ARID1A (c.2988 + 1G> A) associada à síndrome de Coffin‐Siris tipo 2 (autossômica dominante), OMIM 614607.
A síndrome de Coffin‐Siris é uma síndrome de malformação congênita rara, da qual foram descritos menos de 200 casos, e é causada por mutações em vários genes que codificam componentes do complexo BRG1/BRM associated factor (BAF), com 12 subtipos diferentes dependendo da mutação genética, incluindo (da maior para a menor proporção de casos) ARID1B, SMARCB, SMARCA4, ARID1A, SOX11, SMARCE1 e PHF6.1,2 O complexo BAF é um remodelador de cromatina dependente de ATP e está envolvido na transcrição e diferenciação celular e reparo do DNA; parece haver uma correlação fenótipo‐genótipo, porque mutações no BAF têm sido relacionadas a anormalidades no cabelo, unhas e quirodáctilos. É uma síndrome clinicamente heterogênea, cujos principais sinais incluem atraso cognitivo ou de desenvolvimento discreto a grave, características faciais grosseiras e hipoplasia ou aplasia ungueal e da falange distal do quinto quirodáctilo e, ocasionalmente, de quirodáctilos adicionais (os pododáctilos geralmente são afetados em indivíduos com envolvimento de múltiplos dedos). Essas características faciais distintas incluem sobrancelhas grossas e cílios longos, ponte nasal ampla, boca larga com lábios grossos e evertidos e posição anormal do pavilhão auricular. Outras características menores incluem hipotonia, hirsutismo ou hipertricose e cabelos escassos no couro cabeludo, baixa estatura, dificuldades de alimentação, crescimento lento e anomalias congênitas, incluindo microcefalia, manifestações oftalmológicas e malformações cardíacas, gastrintestinais, geniturinárias e do sistema nervoso.1,3,4
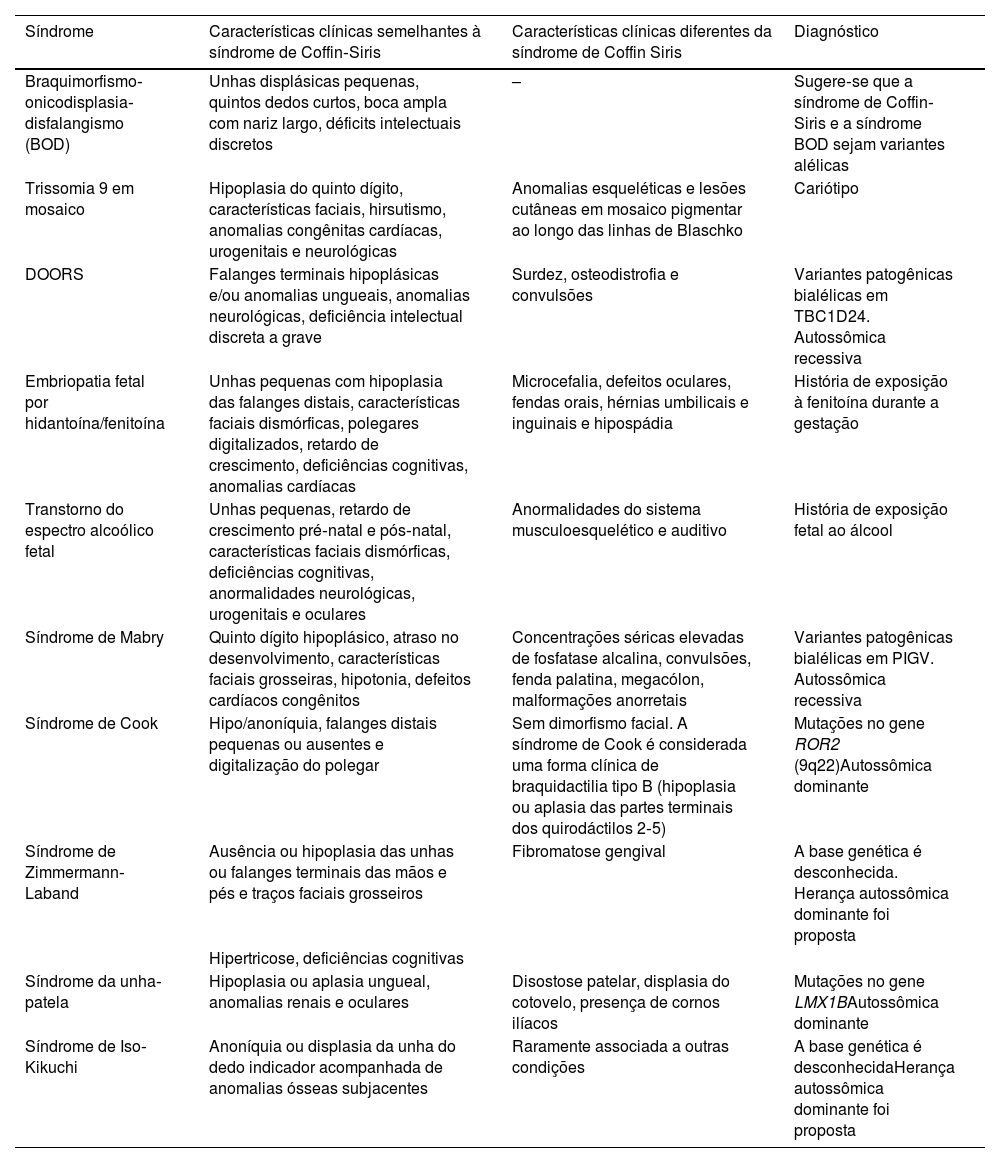
O diagnóstico diferencial inclui síndrome de braquimorfismo‐onicodisplasia‐disfalangismo (BOD), trissomia 9 em mosaico, síndrome DOORS (surdez, onicodistrofia, osteodistrofia, deficiência intelectual), embriopatia fetal por hidantoína/fenitoína, distúrbios do espectro alcoólico fetal, síndrome de Mabry, síndrome de Cook, síndrome de Zimmermann‐Laband, síndrome unha‐patela e síndrome de Iso‐Kikuchi. A tabela 1 resume as semelhanças e diferenças clínicas desses diagnósticos diferenciais em relação à síndrome de Coffin‐Siris, cujo diagnóstico definitivo é genético.3
Diagnóstico diferencial da síndrome de Coffin‐Siris
| Síndrome | Características clínicas semelhantes à síndrome de Coffin‐Siris | Características clínicas diferentes da síndrome de Coffin Siris | Diagnóstico |
|---|---|---|---|
| Braquimorfismo‐onicodisplasia‐disfalangismo (BOD) | Unhas displásicas pequenas, quintos dedos curtos, boca ampla com nariz largo, déficits intelectuais discretos | – | Sugere‐se que a síndrome de Coffin‐Siris e a síndrome BOD sejam variantes alélicas |
| Trissomia 9 em mosaico | Hipoplasia do quinto dígito, características faciais, hirsutismo, anomalias congênitas cardíacas, urogenitais e neurológicas | Anomalias esqueléticas e lesões cutâneas em mosaico pigmentar ao longo das linhas de Blaschko | Cariótipo |
| DOORS | Falanges terminais hipoplásicas e/ou anomalias ungueais, anomalias neurológicas, deficiência intelectual discreta a grave | Surdez, osteodistrofia e convulsões | Variantes patogênicas bialélicas em TBC1D24. Autossômica recessiva |
| Embriopatia fetal por hidantoína/fenitoína | Unhas pequenas com hipoplasia das falanges distais, características faciais dismórficas, polegares digitalizados, retardo de crescimento, deficiências cognitivas, anomalias cardíacas | Microcefalia, defeitos oculares, fendas orais, hérnias umbilicais e inguinais e hipospádia | História de exposição à fenitoína durante a gestação |
| Transtorno do espectro alcoólico fetal | Unhas pequenas, retardo de crescimento pré‐natal e pós‐natal, características faciais dismórficas, deficiências cognitivas, anormalidades neurológicas, urogenitais e oculares | Anormalidades do sistema musculoesquelético e auditivo | História de exposição fetal ao álcool |
| Síndrome de Mabry | Quinto dígito hipoplásico, atraso no desenvolvimento, características faciais grosseiras, hipotonia, defeitos cardíacos congênitos | Concentrações séricas elevadas de fosfatase alcalina, convulsões, fenda palatina, megacólon, malformações anorretais | Variantes patogênicas bialélicas em PIGV. Autossômica recessiva |
| Síndrome de Cook | Hipo/anoníquia, falanges distais pequenas ou ausentes e digitalização do polegar | Sem dimorfismo facial. A síndrome de Cook é considerada uma forma clínica de braquidactilia tipo B (hipoplasia ou aplasia das partes terminais dos quirodáctilos 2‐5) | Mutações no gene ROR2 (9q22)Autossômica dominante |
| Síndrome de Zimmermann‐Laband | Ausência ou hipoplasia das unhas ou falanges terminais das mãos e pés e traços faciais grosseiros | Fibromatose gengival | A base genética é desconhecida. Herança autossômica dominante foi proposta |
| Hipertricose, deficiências cognitivas | |||
| Síndrome da unha‐patela | Hipoplasia ou aplasia ungueal, anomalias renais e oculares | Disostose patelar, displasia do cotovelo, presença de cornos ilíacos | Mutações no gene LMX1BAutossômica dominante |
| Síndrome de Iso‐Kikuchi | Anoníquia ou displasia da unha do dedo indicador acompanhada de anomalias ósseas subjacentes | Raramente associada a outras condições | A base genética é desconhecidaHerança autossômica dominante foi proposta |
DOORS, surdez, onicodistrofia, osteodistrofia, deficiência intelectual.
O manejo dos pacientes com diagnóstico de síndrome de Coffin‐Siris é sintomático e consiste em terapias ocupacionais, físicas e alimentares, incluindo suplementação nutricional e/ou colocação de sonda de gastrostomia conforme necessário. O prognóstico depende da extensão do envolvimento.
A avaliação anual por diferentes especialistas é necessária, como otorrinolaringologista, oftalmologista, neurologista e/ou gastroenterologista para avaliar o progresso do desenvolvimento e as intervenções terapêuticas e educacionais necessárias.
A síndrome de Coffin‐Siris é uma síndrome clinicamente heterogênea. Embora o envolvimento das unhas e a hipoplasia dos dedos possam estar entre os sinais clínicos menos graves, os dermatologistas devem estar familiarizados com essas manifestações, que muitas vezes são fundamentais para estabelecer o diagnóstico.
Suporte financeiroNenhum.
Contribuição dos autoresAlba Navarro‐Bielsa: Concepção e planejamento do estudo; obtenção dos dados ou análise e interpretação dos dados; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; aprovação da versão final do manuscrito.
Daniel Ruiz Ruiz‐de‐Larramendiz: Concepção e planejamento do estudo; obtenção dos dados ou análise e interpretação dos dados; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Pilar Abenia‐Usón: Concepção e planejamento do estudo; obtenção dos dados ou análise e interpretação dos dados; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; aprovação da versão final do manuscrito.
Tamara Gracia‐Cazaña: Concepção e planejamento do estudo; obtenção dos dados ou análise e interpretação dos dados; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; aprovação da versão final do manuscrito.
Yolanda Gilaberte: Concepção e planejamento do estudo; obtenção dos dados ou análise e interpretação dos dados; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Navarro‐Bielsa A, Ruiz‐de‐Larramendiz DR, Abenia‐Usón P, Gracia‐Cazaña T, Gilaberte Y. Nail dysplasia and digital hypoplasia ‐ Coffin‐Siris syndrome. An Bras Dermatol. 2024;99:749–52.
Trabalho realizado no Hospital Miguel Servet, Saragoça, Espanha
