






O complexo da esclerose tuberosa (CET) é síndrome neurocutânea multissistêmica com fenótipos variados. A atualização recente dos critérios diagnósticos do CET reafirmou o critério diagnóstico genético definido como o achado de uma alteração patogênica no DNA nos genes TSC1 ou TSC2. A atualização também modificou ligeiramente os critérios diagnósticos clínicos finais. Lesões cutâneas associadas ao CET na infância são sinais clínicos importantes para selecionar indivíduos com possível CET para seguimento clínico mais detalhado e testes genéticos.
ObjetivoAumentar o conhecimento dos critérios diagnósticos atualizados do CET; avaliar a frequência de lesões cutâneas em pacientes com CET, bem como a primeira apresentação dermatológica; e associar os achados às alterações do TSC1 ou TSC2.
MétodosEstudo observacional transversal. Os dados clínicos e genéticos foram coletados retrospectivamente de 37 pacientes com CET em hospital universitário brasileiro. Pacientes com sinais cutâneos foram examinados e avaliados prospectivamente por 12 meses.
ResultadosAs lesões cutâneas mais precoces foram máculas hipomelanóticas; que juntamente com os angiofibromas também foram as lesões dermatológicas mais frequentes. A razão total de alterações patogênicas no DNA entre os genes TSC2 e TSC1 foi de 8:1. A frequência de uma variante patogênica do TSC2 foi dez vezes maior na presença de fibromas ungueais.
Limitações do estudoAmostra pequena e número limitado de pacientes com variantes patogênicas do TSC1.
ConclusãoOs médicos devem ter conhecimento sobre os critérios diagnósticos atualizados do CET. Os pacientes devem ser monitorados por equipe multidisciplinar e tratados adequadamente. A detecção precoce de lesões cutâneas é importante para o diagnóstico de CET. É descrita associação significante entre alterações patogênicas do gene TSC2 e fibromas ungueais.
O complexo da esclerose tuberosa (CET) é síndrome neurocutânea com fenótipos variados e herança autossômica dominante. A doença é causada por alterações patogênicas no DNA dos genes TSC1 (OMIM 605284) ou TSC2 (OMIM 191092) e é caracterizada por tumores benignos (hamartomas) em órgãos distintos, principalmente cérebro, pele, coração, rins e pulmões.1,2 O gene TSC1 (9q34) codifica a hamartina, e o TSC2 (16p13.3) codifica a tuberina. Sob condições fisiológicas, as proteínas hamartina, tuberina e TBC1D7 (OMIM 612655) formam um complexo citoplasmático que suprime o crescimento celular ao inibir o alvo mecanístico da rapamicina (mTOR).1,3,4 A perda de função bialélica de TSC1 ou TSC2 prejudica o efeito inibitório na via mTOR causando diferenciação e crescimento celular anormais, resultando em lesões hamartomatosas (tabela 1).3‐5
Critérios maiores e menores para o diagnóstico clínico de CET de acordo com Northrup et al., 20213
| Critérios maiores | Critérios menores |
|---|---|
| Máculas hipomelanóticas (≥ 3, com pelo menos 5 mm de diâmetro) | Lesões em confete |
| Angiofibromas faciais (≥ 3) ou placa fibrosa cefálica | Depressões no esmalte dentário (≥ 3) |
| Fibromas ungueais (≥ 2) | Fibromas intraorais (≥ 2) |
| Placas de Shagreen | Mancha acrômica na retina |
| Hamartomas múltiplos na retina | Múltiplos cistos renais |
| Múltiplos tubérculos corticais e/ou linhas de migração radial | Hamartomas não renais |
| Nódulos subependimários (≥ 2) | Lesões ósseas escleróticas |
| Astrocitoma subependimário de células gigantes | |
| Rabdomioma cardíaco | |
| Linfangioleiomiomatosea | |
| Angiomiolipomas (≥ 2)a |
Os termos sublinhados correspondem a critérios atualizados.3
A combinação desses dois critérios principais sem outras características não permite um diagnóstico definitivo.
Diagnóstico clínico definitivo de CET: dois critérios maiores ou um critério maior mais dois ou mais critérios menores.
Possível diagnóstico clínico de CET: um critério maior ou pelo menos dois critérios menores.
Desde 2012, a presença de alteração patogênica no gene TSC1 ou TSC2 é critério definitivo e suficiente para diagnosticar o CET, independentemente dos achados clínicos.4 Em 2021, o critério diagnóstico genético foi confirmado e a combinação de critérios clínicos que levam a diagnósticos clínicos definitivos ou prováveis foi mantida (tabela 1).3 Entretanto, houve poucas alterações nos critérios clínicos. Múltiplos tubérculos corticais e/ou linhas de migração radial substituíram o termo geral displasia cortical como critério maior, e lesões ósseas escleróticas foram restabelecidas como critério menor (tabela 1).3 Pacientes com diagnóstico definitivo, provável ou suspeito de CET devem ser monitorados por equipe multidisciplinar para identificação precoce de sinais e sintomas, manejo clínico e intervenção terapêutica, resultando em melhores desfechos clínicos.3,4
A associação entre genótipos e fenótipos de CET tem sido estudada, destacando‐se as alterações no gene TSC2 mais frequentemente entre pacientes com relatos anteriores de convulsões, presença de síndrome de West (espasmos epilépticos/infantis com início na primeira infância associados a hipsarritmia e regressão do desenvolvimento) e déficit cognitivo mais grave.2 Embora as análises genótipo‐fenótipo se concentrem nos sinais neurológicos e renais, que são as principais causas de morbidade e mortalidade,3 os sinais dermatológicos são uma das apresentações clínicas mais precoces e sua detecção pode levar à suspeita de CET.5,6
O CET tem sido frequentemente subdiagnosticado, principalmente em países em desenvolvimento.7 O aparecimento precoce de alguns sinais cutâneos é altamente sugestivo da doença (tabela 1). Dada sua importância para o diagnóstico e tratamento dessa doença genética neurocutânea, este estudo teve como objetivo caracterizar as lesões cutâneas de 37 pacientes com diagnóstico clínico e genético definitivo de CET. Além disso, os pacientes foram classificados de acordo com o gene alterado; em cada categoria genética verificou‐se a frequência de lesões cutâneas específicas. Nessa coorte de CET, os autores descrevem associação significante entre fibromas ungueais e alterações no gene TSC2.
Pacientes e métodosEste estudo observacional, analítico e transversal foi realizado no Departamento de Pediatria do Complexo Hospitalar de Clínicas da Universidade Federal do Paraná (CHC‐UFPR), Curitiba, Brasil. O protocolo do estudo foi aprovado pelos Comitês de Ética em Pesquisa do CHC/UFPR (CAAE n° 67137317.5.0000.0096) e da Universidade de São Paulo (CAAE n° 12572913.3.3002.5479 e 48259715.2.3003.5505)
Os pacientes incluídos no estudo foram clinicamente diagnosticados com CET de acordo com Northrup et al.4 e tinham uma variante patogênica de DNA previamente identificada por Sanger, amplificação de sonda dependente de ligação multiplex (MLPA, do inglês multiplex ligation‐dependent probe amplification) ou sequenciamento de próxima geração (NGS, do inglês next‐generation sequencing) nos genes TSC1 (NM_000368.4) ou TSC2 (NM_000548.3) na Universidade de São Paulo, São Paulo, Brasil (dados não publicados). Após a publicação dos critérios diagnósticos atualizados,3 os dados foram revisados adequadamente e nenhuma alteração foi feita. Neste estudo ambispectivo, após a família fornecer consentimento informado por escrito, os prontuários médicos foram revisados retrospectivamente. Em seguida, os pacientes foram atendidos prospectivamente pela equipe de Dermatologia Pediátrica durante mutirão para o atendimento de pacientes com CET em outubro de 2021 e outubro de 2022. As informações (identificação, idade, datas, resultado do estudo genético e descrição das lesões cutâneas) foram registradas e as lesões clínicas, fotografadas. Os dados foram analisados estatisticamente no software Excel®.
A amostra foi selecionada de maneira não probabilística e sistemática por conveniência, por ordem de agendamento e admissão no estudo. As medidas de tendência central e dispersão foram expressas como média±desvio padrão para variáveis contínuas com distribuição simétrica e mediana (intervalo interquartil [IQR]) para aquelas com distribuição assimétrica. As variáveis categóricas foram expressas como frequências absolutas e relativas. O teste não paramétrico de Mann‐Whitney foi utilizado para estimar a diferença entre variáveis contínuas com distribuição assimétrica, e os testes exato de Fisher e Qui‐Quadrado de Pearson foram utilizados para estimar a diferença entre variáveis categóricas. Todos os testes consideraram nível de significância mínimo de 5%, erro tipo II de 10% e magnitude do efeito da diferença estimada entre genes mutados de quatro vezes.
ResultadosA amostra do estudo incluiu um total de 37 pacientes (36 pacientes não aparentados), com diagnóstico clínico e genético definitivo de CET, dos quais 20 (54,1%) eram do gênero masculino e 17 (45,9%) do feminino. Todos apresentavam pelo menos um sinal dermatológico considerado critério diagnóstico de CET (tabela 1). Todos os pacientes tiveram a primeira lesão cutânea antes dos 18 anos. A mediana da idade no momento do diagnóstico de CET foi de 18 meses (IIQ: 8‐60), e a mediana da idade na avaliação dermatológica foi de 16 anos (IIQ: 9‐24,5).
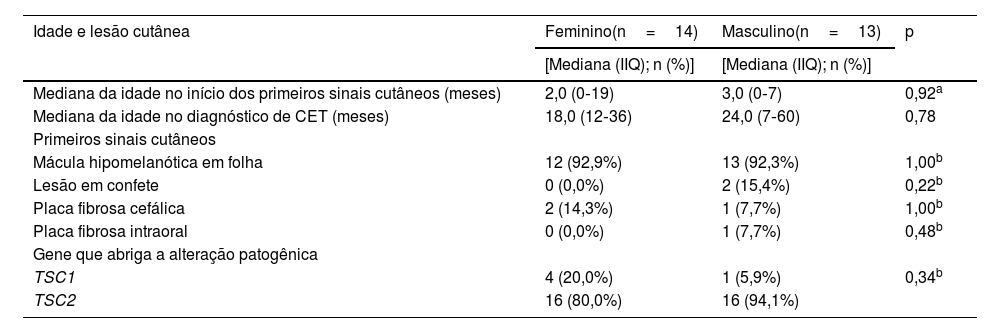
Vinte e sete famílias souberam informar o primeiro sinal cutâneo identificado no paciente. A mediana da idade de início dos pacientes foi de 3 meses (IIQ: 0‐19), e a lesão cutânea mais frequente identificada inicialmente foi a mácula hipomelanótica em forma de folha (25/27; p<0,001). Considerando os pacientes de acordo com o gênero, não foi observada diferença nos primeiros sinais cutâneos, idade de início, idade do diagnóstico de CET ou gene mutado (p>0,05; tabela 2).
Idade na detecção da primeira lesão cutânea, diagnóstico de CET e primeiros sinais cutâneos de acordo com o gênero
| Idade e lesão cutânea | Feminino(n=14) | Masculino(n=13) | p |
|---|---|---|---|
| [Mediana (IIQ); n (%)] | [Mediana (IIQ); n (%)] | ||
| Mediana da idade no início dos primeiros sinais cutâneos (meses) | 2,0 (0‐19) | 3,0 (0‐7) | 0,92a |
| Mediana da idade no diagnóstico de CET (meses) | 18,0 (12‐36) | 24,0 (7‐60) | 0,78 |
| Primeiros sinais cutâneos | |||
| Mácula hipomelanótica em folha | 12 (92,9%) | 13 (92,3%) | 1,00b |
| Lesão em confete | 0 (0,0%) | 2 (15,4%) | 0,22b |
| Placa fibrosa cefálica | 2 (14,3%) | 1 (7,7%) | 1,00b |
| Placa fibrosa intraoral | 0 (0,0%) | 1 (7,7%) | 0,48b |
| Gene que abriga a alteração patogênica | |||
| TSC1 | 4 (20,0%) | 1 (5,9%) | 0,34b |
| TSC2 | 16 (80,0%) | 16 (94,1%) |
As figuras 1 e 2 ilustram alguns dos principais achados cutâneos nos pacientes com CET desta coorte, que foram máculas hipomelanóticas (92,5%), angiofibromas (87,5%), placas cefálicas (80%), lesões semelhantes a confete (67,5%), placas de Shagreen (55%), fibromas ungueais (55%) e fibromas intraorais (25%). Quando cada lesão cutânea do CET foi considerada individualmente, não houve diferença entre os gêneros (p>0,05; fig. 3A). O número de angiofibromas também foi semelhante entre os gêneros (p=0,31). Dados coletados de 37 pacientes (mediana de idade de 16 [4‐41] anos) examinados em dois momentos (outubro de 2021 e outubro de 2022) não revelaram nenhuma nova lesão cutânea um ano depois.
 Máculas hipomelanóticas no abdme (A) e placa de Shagreen no dorso de paciente adolescente (B).")
 Lesões em confete (setas) na região anterior da tíbia (A) e fibromas ungueais no 4° e 5° quirodáctilos de paciente adulto (B).")
 e gene mutado (B). (A) Teste exato de Fisher: máculas hipomelanóticas: p=0,23; angiofibromas: p=1,00; placas cefálicas: p=0,70; lesões em confete: p=0,50; fibromas ungueais: p=0,53; placas de Shagreen: p=0,33; fibromas intraorais: p=0,73. (B) Teste exato de Fisher: máculas hipomelanóticas: p=0,44; angiofibromas: p=1,00; placas cefálicas: p=0,61; lesões em confete: p=0,18; fibromas ungueais: p=0,07; placas de Shagreen: p=0,42; fibromas intraorais: p=1,00.")
Distribuição de frequência das lesões cutâneas por gênero (A) e gene mutado (B). (A) Teste exato de Fisher: máculas hipomelanóticas: p=0,23; angiofibromas: p=1,00; placas cefálicas: p=0,70; lesões em confete: p=0,50; fibromas ungueais: p=0,53; placas de Shagreen: p=0,33; fibromas intraorais: p=0,73. (B) Teste exato de Fisher: máculas hipomelanóticas: p=0,44; angiofibromas: p=1,00; placas cefálicas: p=0,61; lesões em confete: p=0,18; fibromas ungueais: p=0,07; placas de Shagreen: p=0,42; fibromas intraorais: p=1,00.
Todos os 37 pacientes com CET tiveram DNA avaliado por sequenciamento, incluindo dois irmãos. As 36 alterações patogênicas do DNA foram distribuídas entre o gene TSC1 em quatro indivíduos não aparentados (11,1%) e TSC2 em 32 (88,9%) pacientes, definindo razão de alteração TSC2:TSC1 de 8:1. Havia três variantes de frameshift e uma grande deleção no TSC1. Os dois irmãos apresentavam alteração patogênica do TSC1, totalizando cinco indivíduos com alterações nesse gene. As alterações do TSC2 compreenderam sete variantes de frameshift, sete de missense único, seis nonsense único, três splicing e três variantes de deleção in‐frame, além de três grandes deleções e uma grande duplicação. Uma variante nonsense do TSC2 foi encontrada em três pacientes não aparentados, totalizando 32 pacientes com alterações no TSC2.
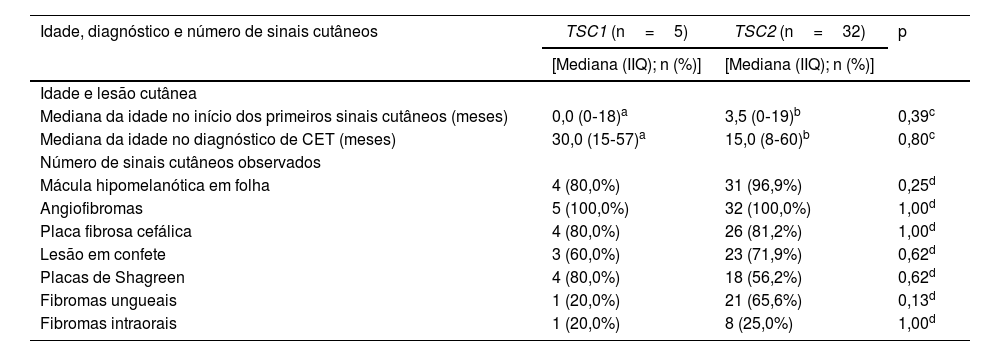
As frequências de alterações patogênicas nos genes TSC1 e TSC2 foram semelhantes entre os gêneros (tabela 2; p=0,34). A idade de início da lesão cutânea e a idade do diagnóstico de CET estavam disponíveis para cinco e 22 pacientes com alterações patogênicas no DNA dos genes TSC1 e TSC2, respectivamente. Não houve diferenças significantes entre os grupos de pacientes com genes alterados distintos, quando as comparações foram feitas para idade no aparecimento da primeira lesão cutânea, no diagnóstico de CET ou tipo da primeira lesão cutânea (tabela 3; p>0,05).
Idade na detecção da primeira lesão cutânea, diagnóstico de CET e sinais cutâneos totais de acordo com o gene que abriga a alteração patogênica
| Idade, diagnóstico e número de sinais cutâneos | TSC1 (n=5) | TSC2 (n=32) | p |
|---|---|---|---|
| [Mediana (IIQ); n (%)] | [Mediana (IIQ); n (%)] | ||
| Idade e lesão cutânea | |||
| Mediana da idade no início dos primeiros sinais cutâneos (meses) | 0,0 (0‐18)a | 3,5 (0‐19)b | 0,39c |
| Mediana da idade no diagnóstico de CET (meses) | 30,0 (15‐57)a | 15,0 (8‐60)b | 0,80c |
| Número de sinais cutâneos observados | |||
| Mácula hipomelanótica em folha | 4 (80,0%) | 31 (96,9%) | 0,25d |
| Angiofibromas | 5 (100,0%) | 32 (100,0%) | 1,00d |
| Placa fibrosa cefálica | 4 (80,0%) | 26 (81,2%) | 1,00d |
| Lesão em confete | 3 (60,0%) | 23 (71,9%) | 0,62d |
| Placas de Shagreen | 4 (80,0%) | 18 (56,2%) | 0,62d |
| Fibromas ungueais | 1 (20,0%) | 21 (65,6%) | 0,13d |
| Fibromas intraorais | 1 (20,0%) | 8 (25,0%) | 1,00d |
Os fibromas ungueais foram mais frequentes entre os pacientes com alteração genética no TSC2 do que no TSC1 (fig. 3B; 65,6% vs. 20,0%, para TSC2vs. TSC1; p=0,07). As razões de chances (odds ratio) estimadas para lesões cutâneas específicas revelaram um aumento de dez vezes para fibroma ungueal quando uma alteração patogênica de TSC2 estava presente (fig. 4A; razão de chances=10,50; intervalo de confiança de 95%: 1,12‐67,21, p=0,03). O número de angiofibromas por paciente foi semelhante entre pacientes portadores de alteração patogênica em diferentes genes (fig. 3B; p=0,64) e variou de zero a cinco (fig. 4B).
Discussão e odds ratio de lesões cutâneas de CET para a mutação TSC2 (B). (A) Teste Qui‐Quadrado de Pearson: p=0,64. (B) Fibromas ungueais: OR=10,50; p=0,03. OR, odds ratio.")
As lesões cutâneas associadas ao CET devem ser reconhecidas pelos clínicos gerais e pediatras, pois são comumente os primeiros sinais perceptíveis dessa doença e compreendem quatro dos 11 critérios maiores e três dos sete critérios menores para o diagnóstico clínico de CET (tabela 1). O diagnóstico precoce é importante para o acompanhamento longitudinal e o início do tratamento adequado, limitando o crescimento das lesões e a morbidade relacionada. Os inibidores de mTOR receberam aprovação clínica como terapia oral para lesões específicas de TSC, como por exemplo o everolimus para tratar astrocitoma subependimário de células gigantes,8 angiomiolipoma renal,9 e como tratamento adjuvante de convulsões de início parcial do CET;10 e sirolimus (rapamicina) para linfangioleiomiomatose.11 Em estudos clínicos, o sirolimus tópico diminuiu os angiofibromas, as máculas hipomelanóticas e as placas cefálicas e é bem tolerado por crianças.12‐14 Portanto, as lesões cutâneas do CET não são importantes apenas para o diagnóstico da doença, mas podem ser tratadas de maneira eficaz através da hiperativação do mTOR.
Em nossa coorte de 37 pacientes com diagnóstico clínico e genético definitivo de CET, os sinais cutâneos iniciais mais comuns foram máculas hipomelanóticas, que também foram a apresentação dermatológica mais frequente (92,5%). Máculas hipomelanóticas ocorrem em aproximadamente 90% dos pacientes e têm início geralmente ao nascimento ou na primeira infância,6 como observado (n=27 casos informados; idade média de 3 meses; tabela 2). Outros primeiros sinais cutâneos menos observados foram lesões em confete, placas fibrosas cefálicas ou intraorais (tabela 2). Entre os bebês de 1 mês de vida com pelo menos um sinal sugestivo de CET, as máculas hipomelanóticas foram a causa da suspeita diagnóstica em 31% deles.15 Em pacientes com até 4 meses de vida, máculas hipomelanóticas foram o sinal inicial em 35%.7 O presente estudo não avaliou sinais não dermatológicos. Ele confirma que, entre as lesões cutâneas, as máculas hipomelanóticas são o primeiro sinal mais frequente em pacientes com CET (tabela 2).
Os angiofibromas faciais foram a segunda lesão mais frequente (87,5%), seguidos pelas placas cefálicas (80%). A ocorrência geral estimada de angiofibroma (75%)6 aumenta na segunda década de vida, após a puberdade.7 Assim, a frequência dos angiofibromas depende da média de idade dos pacientes no momento da admissão na coorte. As placas de Shagreen são geralmente observadas a partir da primeira década de vida, mais comumente a partir dos 5 anos,5,16 em frequência (50%) semelhante à observada pelos autores do presente estudo (55%). Lesões semelhantes a confete foram detectadas em 67,5% dos pacientes. Em geral, começam na primeira década de vida, mas têm utilidade limitada como critério diagnóstico em adultos, pois podem se desenvolver como consequência de exposição solar crônica.6 Uma história de início na primeira infância é sugestiva de CET.5 Os fibromas intraorais ocorrem em 20‐50% dos pacientes, mais frequentemente em adultos do que em crianças.6 Essas lesões foram detectadas em 10 pacientes desta coorte com CET (25%). A distribuição das lesões cutâneas não foi diferente entre os gêneros (tabela 2). Os fibromas gengivais foram identificados com mais frequência no gênero masculino do que no feminino.17
A média de idade ao diagnóstico foi de 18 meses na presente amostra, 15 meses depois da média de idade na detecção do primeiro sinal cutâneo e mais tarde do que a média de idade ao diagnóstico em outro estudo recente (n=86; seis meses) que adotou os mesmos critérios diagnósticos.4 Nesse estudo alemão, o rabdomioma cardíaco, um critério clínico maior de CET (tabela 1), detectado por imagens fetais pré‐natais, contribuiu para o diagnóstico precoce da doença em 22% dos casos.6 Parece que os pacientes da presente coorte brasileira têm acesso heterogêneo a exames de imagem fetais e dependem mais comumente de hospitais terciários de referência para o estabelecimento do diagnóstico de CET.
O teste genético é útil para confirmar o diagnóstico de CET, particularmente em casos clinicamente desafiadores em estágios iniciais. Nessa análise, as variantes patogênicas de TSC1 e TSC2 foram observadas em 13,5% (5/37) e 86,5% (32/37) dos pacientes, respectivamente, razoavelmente acima da proporção observada em outros estudos.17‐21 A predominância de alterações genéticas patogênicas do TSC2, oito vezes mais comuns que as variantes patogênicas do TSC1, foi observada predominantemente no gênero feminino (oito vezes no gênero feminino e 3,4 vezes no masculino; tabela 2). A chance de mutação no TSC2 foi 10 vezes maior na presença de fibromas ungueais, embora esses dados devam ser vistos com cautela em virtude do amplo intervalo de confiança associado ao pequeno número de casos com mutação no TSC1.
Um fenótipo neurológico mais grave pode estar associado à mutação no TSC2,17‐21 enquanto as lesões cutâneas e a associação genotípica parecem ser mais variáveis entre os estudos, exceto para angiofibromas faciais. O banco de dados de história natural do TSC revelou associação significante entre a mutação do TSC2 e angiofibromas.22 O grau médio de angiofibroma facial parece ser maior em pacientes com alteração de TSC2 do que de TSC1.18 Não foi observada diferença no número de angiofibromas, o que pode ser devido ao pequeno número de pacientes com mutação no TSC1, embora pareça haver tendência ao aumento do número de angiofibromas na presença de mutação no gene TSC2 (fig. 4A). Em outro estudo, crianças de 2 anos com mutação no gene TSC1 apresentaram menos máculas hipomelanóticas e angiofibromas faciais do que aquelas com alteração genética no TSC2.23 Por outro lado, placas de Shagreen foram observadas com mais frequência em pacientes com alteração no gene TSC1 do que no TSC2.17 Entre 38 pacientes com CET com pelo menos um sinal cutâneo, o exame dermatológico por si só foi suficiente para estabelecer o diagnóstico definitivo da doença em 34 deles, demonstrando a importância do reconhecimento precoce das lesões cutâneas.24,25
A presente coorte de CET de um centro clínico de referência no sul do Brasil revela a associação entre fibromas ungueais e alteração genética do gene TSC2. Uma limitação do estudo é o pequeno tamanho amostral e o número limitado de pacientes com variantes patogênicas do TSC1. Mais estudos são necessários para avaliar a associação da apresentação dermatológica do CET com o genótipo. Foi confirmada a importância da detecção de lesões cutâneas para identificação precoce do CET. Os médicos devem estar cientes das possíveis lesões mucocutâneas do CET, sua expressividade variável e da história natural da doença.26
Suporte financeiroEssa pesquisa recebeu suporte financeiro dos subsídios 2013/08028‐1 e 2019/10868‐4da Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) e CAPES (Processo: 88881.132401/2016‐01; Brasil).
Contribuição dos autoresBeatriz Azevedo Nunes: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; análise estatística; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Ana Karolina Ferreira Gonçalves Romano: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; análise estatística; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Mariana Aparecida Pasa Morgan: Concepção e planejamento do estudo; análise e interpretação dos dados; análise estatística; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Alice Andrade Gonçalves: Obtenção de dados; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Laís Faria Masulk Cardozo: Concepção e planejamento do estudo; obtenção de dados; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Luiz Gustavo Dufner de Almeida: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; análise estatística; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Luciana Amaral Haddad: Concepção e planejamento do estudo; Análise e interpretação dos dados; análise estatística; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Ana Chrystina de Souza Crippa: Concepção e planejamento do estudo; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Sergio Antonio Antoniuk: Concepção e planejamento do estudo; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Kerstin Taniguchi Abagge: Concepção e planejamento do estudo; Análise e interpretação dos dados; análise estatística; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; e aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Nenhum.
Como citar este artigo: Nunes BA, Romano AKFG, Morgan MAP, Gonçalves AA, Cardozo LFM, de Almeida LGD, et al. A dermatological assessment of pediatric patients with tuberous sclerosis complex (TSC). An Bras Dermatol. 2024;99:662–9.
Trabalho realizado no Complexo Hospitalar de Clínicas, Universidade Federal do Paraná, Curitiba, PR, Brasil.
