













A pele, ao refletir processos internos, exterioriza o que acontece no organismo em muitas doenças. Nesse sentido, ela, como órgão, extrapola as suas funções de proteção, barreira, e sinaliza a existência de enfermidades sistêmicas, ampliando a importância do médico dermatologista para além da superfície cutânea. O dermatologista, por conseguinte, investiga hipóteses diagnósticas de afecções relacionadas a todos os sistemas e referencia o paciente para a especialidade adequada. Além do exame clínico por olhar treinado, a pele, por seu fácil acesso, ainda é o local ideal para realização de biópsias, que muitas vezes esclarecem o diagnóstico. Este manuscrito é a segunda parte do artigo sobre manifestações cutâneas das doenças sistêmicas. Na primeira parte foram descritas as manifestações cutâneas das principais doenças reumatológicas, das doenças granulomatosas e abordamos também as manifestações vasculares. No presente, abordaremos como as doenças metabólicas, cardiovasculares, renais e gastrointestinais podem se manifestar no tegumento. Discutiremos também as doenças oncológicas com suas repercussões cutâneas. Será discutido o prurido e sua correspondência clínica cutânea. Por fim, será apresentada atualização sobre sinais cutâneos da infecção pelo coronavírus SARS‐CoV2.
A pele se comunica com o organismo, muitas vezes deixando transparecer os primeiros sinais e/ou sintomas de uma doença interna – essas podem ser, inclusive, as únicas expressões de doenças sistêmicas.
Nesta segunda parte de nosso trabalho, abordamos de maneira abrangente as manifestações cutâneas das doenças metabólicas, cardiovasculares, renais, gastrintestinais e oncológicas. Em algumas dessas doenças, é possível separar as manifestações cutâneas em específicas (a histopatologia representa e sela o diagnóstico da doença investigada) e inespecíficas da doença (quando na lesão cutânea não há características histopatológicas da doença sistêmica). Em outras, essa classificação não é aplicável. Descreveremos essa divisão, quando possível. Discutiremos também o prurido. Por fim, apresentaremos uma revisão atual das manifestações cutâneas da infecção pelo novo coronavírus SARS‐CoV‐2, visto sua relevância no contexto pandêmico atual.
Doenças metabólicasDiabetes mellitus1. Necrobiose lipoídica: doença inflamatória crônica não pruriginosa, granulomatosa com degeneração do colágeno. Habitualmente, inicia‐se com pápulas eritematosas e evolui lentamente para placa marrom‐amarelada com centro atrófico e telangiectasias. Há maior prevalência em mulheres do que em homens, e é comumente localizada na região anterior das pernas com localização simétrica. Em 35% dos casos, as lesões podem ulcerar, comumente complicadas por infecções bacterianas secundárias. A maioria dos casos tem curso crônico, porém algumas vezes pode ocorrer melhora espontânea.1
2. Scleredema adultorum de Buschke: caracterizado por endurecimento extenso lentamente progressivo, semelhante à esclerodermia da pele do dorso, região cervical posterior, ombros e face. É definida por deposição de colágeno e mucopolissacarídeos na derme, com espessamento da pele, rigidez e comprometimento da motilidade, principalmente nos ombros. É desordem rara, que pode ser sub‐reconhecida. Também pode ser encontrado em outras doenças como neoplasias, paraproteinemias e infecções. Pápulas de Huntley (pequenas pápulas eritematosas agrupadas) podem estar presentes. Na doença extensa, toda a pele, bem como órgãos internos, como pulmão, podem estar envolvidos.1
3. Bulose diabeticorum: geralmente em pacientes com diabetes de longa data (também pode ser manifestação inicial da doença), que se manifesta com bolhas serosas tensas sem sinais de inflamação cutânea e indolores. Surgem predominantemente nas extremidades inferiores, especialmente nos pés; entretanto, podem acometer as mãos e o tronco. As lesões emergem rapidamente e cicatrizam em algumas semanas. Cerca de 0,5% dos indivíduos com diabetes desenvolvem bolhas específicas no curso da doença.1
4. Granuloma anular (GA): caracteriza‐se por pápulas normocrômicas a eritematosas, formando placas de conformação anular no dorso dos pés e mãos ou na superfície extensora de articulações (como o cotovelo). Costuma aparecer em diabéticos, mas a associação com doenças infecciosas, como hepatite e HIV, e tumores, como linfoma e carcinoma, também é descrita. A doença é frequentemente assintomática e as lesões involuem no centro da placa com hipo ou hiperpigmentação. A maioria das lesões cicatriza espontaneamente. Formas disseminadas de GA são cosmeticamente inestéticas e podem ser algo pruriginosas1 (fig. 1).

5. Acantose nigricans: caracteriza‐se por placas hiperpigmentadas intertriginosas. Região cervical e axilas são comumente afetadas no paciente diabético. Em geral, antecede o diagnóstico de diabetes e pode aparecer no estado de hiperinsulinemia com valores normais de hemoglobina glicosilada. Também pode ser observada na síndrome dos ovários policísticos e em obesos. Pode se manifestar como doença paraneoplásica e em doenças gastrintestinais malignas. Além disso, casos induzidos por fármacos foram relatados.1
6. Vitiligo: como doença autoimune, ocorre concomitante a outras doenças autoimunes com frequência, como diabetes mellitus insulinodependente e doenças da tireoide. As lesões acrômicas frequentemente afetam as extremidades, face e região cervical, bem como tronco, e tendem a ser simétricas e de fácil diagnóstico.1
7. Carotenodermia: pele e unhas amareladas podem ser atribuídas à hipercarotenemia em diabéticos.1
8. Líquen plano: cerca de 25% dos pacientes com líquen plano têm diabetes. Os achados clínicos mostram lesões característica acompanhadas de prurido localizadas nos tornozelos e punhos, com envolvimento opcional da mucosa. Outras doenças sistêmicas também podem estar associadas ao líquen plano, como enfermidades hepáticas, intestinais e timoma.1
9. Colagenose reativa perfurante adquirida: doença cutânea rara, vista em pacientes diabéticos, bem como na doença renal crônica e hiperuricemia. A patogênese permanece desconhecida. Embora seja rara, a clínica é típica: pápulas e nódulos eritematosos pruriginosos umbilicados que representam a eliminação transepidérmica de debris dérmicos. O fenômeno de Koebner é comum nesses pacientes (fig. 2).1
. Arquivo pessoal, Dr. Alexandre Gripp.")
10. Angiopatia diabética e doenças de pele associadas à neuropatia:a) Síndrome do pé diabético: a combinação de angiopatia diabética, neuropatia e trauma mecânico desempenha um papel importante na sua patogênese, que cursa com úlcera crônica, na qual 25% dos pacientes desenvolvem complicações como infecções e osteomielite.1b) Síndrome da mão diabética: o diabetes também pode provocar distúrbios musculoesqueléticos nas mãos, como limitação da mobilidade articular (LMA), doença de Dupuytren (DD) (fig. 3) e síndrome do túnel do carpo (STC). A LMA é caracterizada por espessamento e rigidez do tecido conjuntivo periarticular das pequenas articulações da mão, causando limitação da extensão dos dedos. A DD é causada por fibrose e consequente espessamento e encurtamento da fáscia palmar, resultando em contraturas de flexão. Já a STC caracteriza‐se por hipo ou disestesias na área de inervação do nervo mediano da mão.1c) Dermopatia diabética: pode resultar de pequenos traumas. É manifestação frequente do diabético e acompanha outras doenças como nefropatia, retinopatia ou neuropatia. A doença tende a aparecer nas extremidades inferiores de homens idosos. O achado clínico mostra máculas eritematosas assintomáticas ou pápulas de rápido crescimento. As lesões têm curso recorrente e melhoram espontaneamente, com formação de cicatrizes hipotróficas acastanhadas.1

11. Infecções cutâneas comuns em pacientes com diabetes: manifestações cutâneas de infecções bacterianas e fúngicas são frequentes nos diabéticos. Infecções cutâneas recorrentes devem ser motivo de investigação de diabetes mellitus.
O Staphylococcus aureus pode causar piodermites (foliculite, abcessos e impetigo). Já os estreptococos estão associados a ectima, celulite e impetigo contagioso estreptocócico (neste caso, Streptococcus – hemolítico). A fasciíte necrosante é complicação rara da infecção por Streptococcus; porém, S. aureus ou bactérias anaeróbias podem estar envolvidas. A gangrena de Fournier é variante perineal ou genitoanal grave da fasciíte necrosante com elevada taxa de mortalidade.
A pele do trato otorrinolaringológico também é propensa a infecções bacterianas nos diabéticos. A otite externa maligna é infecção bacteriana invasiva comumente causada por Pseudomonas aeruginosa e pode ter curso letal. A infecção normalmente começa assintomática e causa otalgia com saída de secreção purulenta. Casos graves podem se estender, atingir o crânio e o sistema nervoso central, manifestando‐se com meningite e cerebrite.
A candidíase é infecção fúngica comum em diabéticos. Candida albicans é o patógeno mais prevalente e pode afetar pele e mucosas, bem como a região periungueal (paroníquia). A infecção causada por dermatófitos (dermatofitose cutânea ou onicomicose) também é comum.
Mucormicose rinocerebral é doença grave, de alta mortalidade e associada à cetoacidose diabética. É causada, principalmente, por espécies do gênero Rhizopus, Rhizomucor e Lichtheimia (previamente denominada Absidia), fungos oportunistas e angioinvasivos. Clinicamente, se inicia como sinusite com secreção nasal serosanguinolenta que evolui para edema periorbital, coloração eritemato‐violácea, com sinais de necrose cutânea, mucosa nasal e oral. Há sinais de toxemia, dor local e febre. Pode haver paralisia facial periférica. A evolução é rápida para grave comprometimento do sistema nervoso central.
12. Complicações da terapia antidiabética. a) Reações cutâneas causadas pela insulina: reações alérgicas sistêmicas: angioedema, urticária generalizada ou anafilaxia, rubor, prurido palmoplantar ou disseminado. As reações locais são urticariformes ou como eczema, com eritema, pápulas e vesículas no local da injeção, acompanhadas de prurido. Pode haver doença do soro‐símile. A lipodistrofia, menos comum que as reações alérgicas, ocorre após 6 a 24 meses de aplicação contínua de doses de insulina sempre no mesmo local. A introdução de insulinas purificadas reduziu a incidência de lipoatrofia.1b) Reações cutâneas induzidas por antidiabéticos orais: são observados exantema macular, urticária e eritema multiforme. Fotossensibilidade pode ocorrer com a tolbutamida e a clorpropamida. Erupções liquenoides e lesões semelhantes à rosácea podem ocorrer em 1 a 5% dos indivíduos em uso de hipoglicemiantes orais. De todos os medicamentos hipoglicemiantes orais, as sulfonilureias são as que mais frequentemente causam farmacodermias (as de segunda geração causam menos efeitos adversos cutâneos do que as de primeira geração).
13. Prurido: pode estar associado ao diabetes, e será discutido adiante.
Tireopatias- 1.
Hipertireoidismo (quadros cutâneos associados estão informados na tabela 1).
Tabela 1.Manifestações cutâneas de hipertireoidismo
Pele quente, úmida e macia Rubor Eritema palmar Hiperidrose Afinamento difuso do couro cabeludo Onicólise Mixedema pré‐tibial Acropatia tireoidiana Prurido generalizado Urticária crônica Adaptado de Lause M et al.2
- 2.
Hipotireoidismo (quadros cutâneos associados estão informados na tabela 2).
Tabela 2.Manifestações cutâneas de hipotireoidismo
Pele seca Pele fria e manchada Carotenemia Mixedema Macroglossia Perda do terço lateral das sobrancelhas Cabelo áspero e quebradiço Adaptado de Lause M et al.2
Outras doenças autoimunes podem estar associadas ao hipotireoidismo. Foram relatadas: dermatite herpetiforme, alopecia areata, vitiligo e urticária autoimune.2
DislipidemiaXantomas- a)
Xantoma tuberoso: surge como pequenas pápulas, moles, amareladas, eritematosas, localizado em áreas de pressão (cotovelos, joelhos, região glútea); são indolores e se agrupam formando lesões tumorais. A presença de xantoma tuberoso sugere elevação do colesterol sérico e LDL, mas também pode ser visto com aumento de triglicerídeos. Tem maior associação com a hiperlipidemia mista tipo III e tipo IIa, maior incidência de doença vascular aterosclerótica e são raramente associados à hipercolesterolemia secundária.3
- b)
Xantoma tendinoso: corresponde a nódulos firmes de tamanhos variados e observados em áreas sobre tendões (geralmente tendões extensores das mãos, joelhos, cotovelos e tendão de Aquiles). Estão associados à hipercolesterolemia familiar ou secundária, assim como xantomatose cerebrotendinosa e beta‐sitosterolemia. Pacientes com xantomas tendinosos têm incidência extremamente alta de doença vascular aterosclerótica.3
- c)
Xantoma eruptivo: de manifestação abrupta e eruptiva, caracteriza‐se por múltiplas pápulas medindo de 1 a 4 mm, amareladas, com halo eritematoso. As lesões podem coalescer formando lesões nódulo‐tumorais. Ocorre mais comumente sobre pontos de pressão e superfícies extensoras dos braços. Mais raramente, podem estar difusamente espalhados pelo tronco ou nas membranas mucosas. Ao contrário dos outros xantomas, o xantoma eruptivo pode ser pruriginoso. Estão ligados à hipertrigliceridemia secundária e, mais raramente, primária (tipos IV, V e I). Pacientes com xantoma eruptivo apresentam risco alto de ocorrência de pancreatite.3
- d)
Xantoma plano: é a manifestação clínica mais frequente, classificada em três tipos: xantelasma, xantoma estriado e xantoma plano. Os xantelasmas são pápulas poligonais, amareladas, localizadas nas pálpebras (mais comumente no canto medial). Pelo menos 50% dos pacientes com xantelasmas não apresentam dislipidemia. Quando lipídios séricos estão anormais (pacientes mais jovens), o colesterol é a fração geralmente elevada. Os pacientes podem também apresentar a córnea arcus como sinal de hipercolesterolemia. Algumas hiperlipoproteinemias secundárias, como colestase, também podem estar associadas com xantelasmas (fig. 4).

O xantoma estriado palmar apresenta‐se como lesões planas, de amarelado a laranja, nas dobras palmares, e que ocorrem apenas em pacientes com níveis elevados de colesterol e triglicerídeos.
Os xantomas planos difusos geralmente cobrem grandes áreas da face, região cervical, tórax e braços em indivíduos com ou sem dislipidemia (hipertrigliceridemia em particular), mas que frequentemente apresentam paraproteinemia, incluindo as relacionadas ao mieloma múltiplo.
Adrenal1. Síndrome de Cushing: corresponde ao conjunto de características clínicas causadas principalmente por hipercortisolemia. As manifestações cutâneas incluem face de lua, corcunda de búfalo, correspondente ao depósito de tecido adiposo na região dorso cervical, região cervical aumentada de diâmetro e curto, consequente ao acúmulo de gordura supraclavicular, exoftalmia, pelo depósito de gordura retro‐orbitária, hematomas espontâneos, cicatrização retardada de feridas, atrofia cutânea, striae distensae, hiperpigmentação, acantose nigricans, além de lesões acneiformes (acne dos corticoides).
2. Doença de Addison/insuficiência adrenal primária: decorre mais comumente por mecanismos autoimunes. As principais manifestações mucocutâneas são hiperpigmentação cutânea difusa, capilar e de mucosas. Também podemos observar perda de pelos axilares e pubianos além de bandas longitudinais hiperpigmentadas de localização ungueal.
3. Feocromocitoma: A única manifestação cutânea significativa do feocromocitoma é o rubor paroxístico mais proeminente na face, tórax e extremidades superiores. Quando os feocromocitomas ocorrem como parte de uma síndrome genética, as manifestações cutâneas dessa síndrome também podem ser aparentes.
HiperpituitarismoAcromegalia (quadros cutâneos associados estão informados na tabela 3).
Manifestações mucocutâneas de acromegalia
| Macroglossia |
| Macroqueilia |
| Hiperplasia gengival |
| Características faciais grosseiras |
| Cutis verticis gyrata |
| Hiperpigmentação |
| Acantose nigricans |
| Hiperidrose |
| Hipertricose |
| Trocas de unhas |
Adaptado de Lause M et al.2
As lesões cutâneas podem ser a primeira indicação para a suspeita diagnóstica. A pele e os tecidos subcutâneos tornam‐se finos, os pelos escassos e a pele pálida ou amarelada. Os sinais de hipopituitarismo podem ser evidentes e pode haver sintomas da deficiência de gonadotrofina, especialmente redução na libido. As manifestações endócrinas do hipopituitarismo variam com o tipo, idade de desenvolvimento e grau da deficiência hormonal.
Doenças cardiovascularesAo contrário de outros órgãos, como fígado (e o trato gastrintestinal em si) e o sistema endócrino, o sistema cardiovascular não apresenta nenhuma doença própria que possa ter manifestações específicas na pele. Existem inúmeras enfermidades que desenvolvem manifestações cardíacas e cutâneas inespecíficas, que podem ser classificadas em genodermatoses com acometimento cutâneo e cardiovascular e sinais cutâneos de doenças sistêmicas inflamatórias ou neoplásicas com manifestações cardiovasculares. São exemplos:
Genodermatoses1. Complexo de Carney: trata‐se de neoplasia endócrina múltipla familial, autossômica dominante. Apresenta pigmentação cutânea e mucosa (lentigos na face, lábios, pálpebras, conjuntiva e mucosa oral), mixomas cardíacos e cutâneos (e também mamários) e tumores endócrinos, que podem ser: adenomas hipofisários produtores de hormônio do crescimento e prolactina e doença adrenocortical nodular pigmentada primária, neoplasia testicular, adenoma ou carcinoma da tireoide e cistos ovarianos.4 Tem como subtipos os acrônimos: LAMB e NAME. LAMB se refere a lentigos, mixomas atriais e nevo azul. NAME significa: nevos, mixoma atrial, neurofibroma mixoide e efélides.
2. Síndrome LEOPARD: acrônimo de lentigos, alteração de condução eletrocardiográfica, hipertelorismo, estenose pulmonar, genitália anormal, retardo do crescimento e surdez sensorial.5
3. Síndrome de Ehlers‐Danlos: caracterizada por diversos defeitos da estrutura do colágeno V, levando a alteração da pele (pseudotumores musculoides, hematomas espontâneos), articulações e parede dos vasos sanguíneos. Em 2017, a doença foi classificada em 13 variantes. A doença pode ter um espectro de manifestação clínica, no qual a doença assintomática, expressa por hiperflexibilidade articular não sindrômica, é o fenótipo mais frequente.6
4. Pseudoxantoma elástico: genodermatose de herança autossômica recessiva por mutações do gene ABCC6 no cromossoma 16, caracterizada pela mineralização (depósito de cálcio e outros minerais) e degeneração das fibras elásticas, que levam a comprometimento cutâneo (placas amareladas pruriginosas na região cervical, axilar e outras flexuras), vascular (estreitamento, oclusão e aneurismas de artérias periféricas ou cerebrais – com claudicação intermitente dos membros inferiores, angina cardíaca ou isquemia/hemorragia cerebral e hemorragia gastrintestinal) e oftalmológica, essa expressa por estrias angioides retinianas, com redução da acuidade visual.7
5. Hemocromatose hereditária: doença de herança autossômica recessiva que causa depósito de ferro nos hepatócitos, fibras musculares do miocárdio e em outras células. As principais manifestações clínicas cutâneas são hiperpigmentação acastanhada a acinzentada da pele com bronzeamento rápido à mínima exposição ao sol (por melanina, que se segue aos depósitos de hemossiderina em áreas fotoexpostas do corpo – principalmente na cabeça, e também na genitália, mamilos, cicatrizes e flexuras). Também são observados: atrofia e xerose ictiosiforme, queda de cabelos e coiloníquia, hiperpigmentação da mucosa oral, redução do esmalte dentário e da saliva. A prática da flebotomia não regride rapidamente a pigmentação da pele.
6. Esclerose tuberosa: causada por mutações com perda de função dos genes supressores tumorais TSC1 ou TSC2. Em dois terços dos casos a mutação é “de novo”, e em um terço é hereditária. Na pele, ocorrem angiofibromas faciais (em 75% dos indivíduos), fibromas periungueais (20 a 80% dos casos) e orais (50% dos casos), placas fibróticas na região frontal (25% dos casos), máculas hipocrômicas (90% dos casos) e “placas shagreen”, que são nevos de tecido conjuntivo e estão presentes em 50% dos pacientes. Atualmente, acredita‐se que os angiofibromas faciais são induzidos pela radiação ultravioleta; é possível que a fotoproteção reduza seu surgimento.8
7. Doença de Fabry: trata‐se de erro inato do metabolismo, no qual não há catabolização dos lipídios. As manifestações dermatológicas se iniciam na infância, com acroparestesias e intolerância ao calor, acompanhados de náuseas, vômitos, dores abdominais e neuropáticas. Durante a vida surgem os angioqueratomas e telangiectasias (localizados na região glútea e nas coxas).9
8. Amiloidose sistêmica: pode ser classificada em hereditária (tipos ATTR, ALys, Ab2M, AFib, AApoA1, AGel) ou adquirida (tipos AL, AA, ATTR, ALect1, Ab2M). A amiloidose AL é a forma mais frequente (associada a discrasias plasmocitárias, mais comumente o mieloma múltiplo) e a que apresenta mais frequentemente manifestações cutâneas (porém, em apenas em um terço dos casos). Podem estar presentes macroglossia e púrpura periorbitária. Atentar para a púrpura periorbitária isolada, que ocorre em outras formas de amiloidose. Além dessas manifestações clínicas, outros achados que indicam necessidade de investigação de amiloidose são: onicodistrofia com lâmina ungueal fina, onicorrexis, pterígio distal e onicosquizia.10 As formas exclusivamente cutâneas da amiloidose por depósito de citoqueratina 5 – CK5 (amiloidose maculosa e líquen amiloidótico) não evoluem para a doença sistêmica.
Doenças inflamatórias e neoplásicas1. Febre reumática: trata‐se de doença autoimune desencadeada por anticorpos contra Streptococcus beta‐hemolítico do grupo A, que na fase aguda além da febre apresenta manifestações cutâneas tipo eritema marginatum, ou seja, lesões em placas de bordas circinadas eritematosas não pruriginosas e centro claro e comprometimento articular migratório.
2. Síndrome carcinoide: manifesta‐se por dor abdominal, diarreia, flushing (face, pescoço e colo), telangiectasias e broncoespasmo devido à liberação de diversas substâncias por tumores neuroendócrinos metastáticos ou extra‐hepáticos (provenientes de células enterocromafíns ou de Kulchitsky). Pode haver sintomas de deficiência de niacina (pelagra) pela utilização do triptofano (precursor da niacina), que é matéria‐prima para produção dos hormônios secretados.11
Fenômenos tromboembólicosFenômenos tromboembólicos de diversas origens têm manifestações cutâneas que são as “pistas” para pensarmos nessas hipóteses diagnósticas. As lesões geralmente se apresentam como petéquias, púrpuras retiformes e hemorragias em estilha. As possíveis causas são síndrome do anticorpo antifosfolipídio e outras coagulopatias. Êmbolos de colesterol podem se apresentar como livedo reticular, púrpura retiforme e eosinofilia. Vasculite leucocitoclástica pode ocorrer no contexto de doenças infecciosas, tais como hepatites, endocardite infecciosa bacteriana ou fúngica e, em certos casos associados à positividade de p‐ANCA. A endocardite infecciosa pode cursar com lesões de Janeway que correspondem a máculas eritematosas a purpúricas nas palmas e plantas, geralmente na base do primeiro e quinto quirodáctilos, e aos nódulos de Osler, que ocorrem como pápula ou nódulos dolorosos, eritemato‐violáceos, nas mesmas localizações das lesões de Janeway.
Outras manifestações cutâneas isoladas como possíveis observações semiológicas de doenças cardíacas1. Baqueteamento digital: sugere doença cardíaca e/ou doença pulmonar crônica, ou mesmo neoplasia pulmonar.
2. Cianose: doença cardíaca cianótica, especialmente crianças com cardiopatias congênitas; metemoglobinemia (esta, além da cianose, pode acompanhar‐se de pele acinzentada), vasoconstrição intensa (síndrome de Raynaud).
3. Prega diagonal no lóbulo da orelha (sinal de Frank): possivelmente relacionada à doença arterial coronariana e menor evidência científica a associa à aterosclerose carotídea.
4. Edema dos membros inferiores: pode ser causado por insuficiência cardíaca congestiva ou insuficiência do ventrículo direito.
Doenças renaisAs manifestações cutâneas das doenças renais podem ser classificadas em:
1. Lesões cutâneas associadas à uremia:
a) Manifestações que apresentam exame histopatológico específico para o diagnóstico, porém não específica da doença renal em si:
Calcinose cutis: complicação tardia da insuficiência renal, ocorrendo em 1% dos indivíduos com doença renal crônica (IRC) em hemodiálise (HD). É do tipo metastática, causada pelo aumento da relação cálcio/fósforo. Clinicamente, as lesões são pápulas, placas ou nódulos endurecidos assintomáticos ou dolorosos que ulceram e eliminam material branco amarelado. Podem ser vistos na radiografia como depósito radiopaco no subcutâneo e na histopatologia como material homogêneo azulado na derme, que se cora pelo von Kossa.
Calcifilaxia: Também ocorre em 1 a 4% dos pacientes com IRC em HD com hiperparatireoidismo secundário e aumento da relação cálcio/fósforo (> 60 a 70mg2/Dl2). Trata‐se de vasculopatia trombótica devido aos depósitos de cálcio na camada média das arteríolas da pele, além de hiperplasia da camada íntima. As lesões são inicialmente semelhantes ao livedo reticular que progridem para placas de púrpura retiforme extremamente dolorosas e simétricas com formação de úlceras e necrose. Apresenta elevada morbidade, geralmente por sepse decorrente de infecção secundária das áreas de necrose. As lesões comumente se localizam nas áreas de maior concentração de tecido adiposo, como a região glútea, abdome e membros inferiores (coxas e pernas). São mais frequentes em mulheres. Acredita‐se que o indivíduo portador de trombofilia (alterações de proteína C e S e presença de anticorpos antifosfolipídeos) tenha maior predisposição ao desenvolvimento da calcifilaxia. Outros fatores são: hipercalcemia, uso de cálcio com ligante de fosfato, reposição de vitamina D, hiperfosfatemia, hiperparatireoidismo, diabetes mellitus, obesidade, uso de corticosteroides sistêmicos, imunossupressão, trauma12 (fig. 5).

Dermatose perfurante adquirida (doença de Kyrle): associada a diabetes mellitus e IRC. Acomete 4,5 a 10% dos indivíduos em HD. As lesões são pápulas ou nódulos hipercrômicos intensamente pruriginosos, com umbilicação e hiperceratose no topo da lesão, com localização nas superfícies extensoras das extremidades, e fazem diagnóstico diferencial com prurigo nodular. Podem também ocorrer no couro cabeludo, tronco e região glútea. São específicas sob o ponto de vista histopatológico: há acantose, rolhas queratóticas (plugs) em formato de taça, eliminação de debris teciduais pela epiderme e infiltrado inflamatório perilesional composto por neutrófilos e linfócitos. Também podem ocorrer outras dermatoses perfurantes adquiridas, como a foliculite perfurante e a colagenose reativa perfurante (fig. 2).13
Fibrose nefrogênica sistêmica: manifestação rara, relacionada a contraste gadolínio (gadodiamida e gadopentetato) usado em ressonâncias magnéticas e associada a predisposição genética (HLA‐A2) e a estado inflamatório que permite a entrada do gadolínio nos tecidos. Nos tecidos, o contraste é fagocitado pelos macrófagos e desencadeia o processo fibrótico. Clinicamente observam‐se placas simétricas, endurecidas, de eritematosas a hipercrômicas, inicialmente pruriginosas, particularmente nas extremidades. Mais tarde apresentam atrofia da pele, perda de pelos e aparência de “casca de laranja”. Pode evoluir com contraturas dos membros e 5% têm evolução fulminante com acometimento de múltiplos órgãos. A biopsia de pele demonstra feixes colágenos espessados com fendas, depósitos de mucina e proliferação de células fusiformes positivas para CD34 e procolágeno tipo I e também células multinucleadas positivas para CD68 e fator VIII. Faz diagnóstico diferencial com outras doenças que cursam com fibrose (escleromixedema, esclerose sistêmica, síndrome eosinofilia‐mialgia, síndrome do óleo tóxico e fasciíte eosinofílica).12
Oxalose: doença rara, compreende duas formas: oxalose primária e secundária. A oxalose primária do tipo I ocorre em 80% dos casos de oxalose primária e é causada por uma deficiência enzimática autossômica recessiva de alanina: glioxilato aminotransferase (AGT) e então não há metabolização do oxalato em glicolato, se acumulando nos tecidos e causando IRC na maioria dos pacientes aos 20 a 30 anos. A tipo II é causada pela deficiência do complexo enzimático glioxilato redutase/hidroxipiruvato redutase. É mais branda que o tipo I, com clínica de nefrolitíase de repetição e raramente evoluindo para IRC. A tipo III é causada pela mutação do gene HOGA1, levando a perda de função da enzima mitocondrial 4‐hidroxi‐2‐oxoglutarato aldolase e se caracteriza clinicamente por nefrolitíase recorrente na infância (na primeira década de vida). A oxalose primária se manifesta por depósitos intravasculares de oxalato nos rins (o que causa a doença renal terminal), miocárdio, ossos e pele, onde se assemelha clinicamente à calcifilaxia, com livedo reticular e púrpura retiforme e evolui com ulceração e necrose.14 As lesões tendem a ser mais superficiais e menos dolorosas que as lesões de calcifilaxia e ser acompanhadas de placas endurecidas semelhantes às da fibrose nefrogênica. (fig. 6) A oxalose secundária é causada por uma grande ingesta de oxalato ou dos seus precursores (espinafre, beterraba, ruibarbo, vitamina C e intoxicação por etilenoglicol), ou maior absorção pelo cólon, por deficiência de piridoxina ou na IRC (pela menor excreção renal). Leva a depósitos extravasculares de oxalato, o que se traduz clinicamente por pápulas similares a milia e nódulos na derme.15 O diagnóstico histopatológico de ambas as formas é idêntico e específico e evidencia os cristais de oxalato de cálcio (estruturas em formato de agulhas, retangulares ou triangulares amarelo‐acastanhadas com arranjo em rosetas, refringentes sob luz polarizada) nos tecidos ou intravascular. A coloração de von Kossa pode demonstrar depósitos irregulares de cálcio. Há escasso infiltrado inflamatório e algumas células gigantes tipo corpo estranho.

Porfiria cutânea tardia: Ocorre em 1 a 18% dos casos de pacientes com IRC em HD, ocorrer por excreção insuficiente das uroporfirinas e seu consequente acúmulo na pele levando à fotossensibilidade. As porfirinas não são bem dialisadas. Excesso de ferro sérico e a hepatite C são fatores que aumentam a predisposição à sua ocorrência. O paciente apresente vesico‐bolhas e erosões em áreas expostas ao trauma, tais como: mãos, pés, antebraços e mesmo na face, que involuem com formação de cicatrizes e milia. Pode haver hipertricose e hiperpigmentação da face. Na histopatologia há fenda subepidérmica, festonamento da derme papilar e infiltrado inflamatório frustro. Na imunofluorescência direta, observa‐se depósitos de IgG e C3 na junção dermoepidérmica e ao redor dos vasos. Há aumento de ferro, ferritina e uroporfirinas séricos, aumento de isocoproporfirinas nas fezes e de uroporfirinas I e III na urina (fig. 7).13

Pseudoporfiria: também em 1 a 18% dos casos. Não há aumento das porfirinas. Pode ser desencadeado por fármacos, tais como: amiodarona, furosemida, tetraciclinas, isotretinoína, naproxeno, ácido nalidíxico e exposição à luz ultravioleta. A clínica é similar à porfiria cutânea tardia, porém sem hipertricose e hiperpigmentação da face.12
b) Alterações não específicas do ponto de vista histopatológico:
Prurido da doença renal (50 a 90% dos casos).
Alterações cutâneas secundárias ao prurido: as escoriações, prurigo nodular, hiper ou hipopigmentações pós‐inflamatórias, líquen simples crônico.
Xerose: 50 a 80% dos pacientes em hemodiálise. A fisiopatogenia do ressecamento da superfície da pele não é bem compreendida e propicia o prurido.
Discromias: por acúmulo de substâncias não dialisáveis, como hemossiderina (pigmentação castanho‐acinzentada), carotenos e urocromos (coloração amarelada) e hormônio estimulantes do melanócito, revelada por pele acastanhada em área fotoexposta.12
Frost urêmico: (1 a 3% dos casos), ocorre com altos níveis séricos de ureia (250 a 300 mg/dL), pela eliminação desta pelo suor e seu acúmulo na superfície da pele após evaporação da água em locais de transpiração.
c) Alterações ungueais: unhas meio‐a‐meio (unhas de Lindsay) ocorrem em 40% dos indivíduos em hemodiálise, são causadas pela azotemia e regridem após o transplante renal; linhas de Muehrcke (dupla linha transversa branca associada à hipoalbuminemia – valor <2,2 g/dL – da síndrome nefrótica); outras alterações citadas: alterações dos capilares da dobra ungueal; pigmentação melânica da lâmina ungueal distal, leuconíquia, coiloníquia e hemorragias em estilha.12
d) Alterações capilares ocorrem em 30 a 50% dos casos: alopecia por eflúvio telógeno, doença crônica ou pela doença de base que levou à IRC. Também são relatadas perda de brilho e descoloração dos fios.
e) Alterações das mucosas: em 50 a 90% dos casos. Xerostomia e hálito urêmico. A macroglossia pode ocorrer no contexto da IRC associada à amiloidose sistêmica.
f) Lesões cutâneas como consequência do transplante renal: pelo uso de medicamentos: corticosteroides e suas consequências a curto e longo prazo, ciclosporina e por imunossupressores.
g) Lesões de pele causadas pela doença primária que levou a IRC: por exemplo: lesões cutâneas associadas ao LES, doenças genéticas (doença de Fabry, Birt‐Hogg‐Dubé, síndrome unha‐patela, esclerose tuberosa, dentre outras).
Doenças gastrintestinaisDoença inflamatória intestinalAs doenças inflamatórias intestinais (DII) são doenças crônicas que apresentam íntima relação com a pele, causando inúmeras repercussões que contribuem para o aumento da morbidade.16
As úlceras orais podem representar lesão por contiguidade da doença intestinal, tendo a histopatologia semelhante. Contudo, podem também ser uma condição reacional às DII. A estomatite aftosa está presente em mais de 7% de todos os pacientes. Podem aparecer múltiplas úlceras dolorosas com bordas eritematosas, periodontite e periestomatite.16
A pioestomatite vegetante é doença eosinofílica rara que leva à formação de pústulas friáveis, úlceras e placas vegetantes amareladas na mucosa oral e em outras mucosas. Quando acomete a pele, passa a ser chamada de piodermatite‐pioestomatite vegetante.16–18
O eritema nodoso (EN) consiste em nódulos eritemato‐violáceos dolorosos mais tipicamente encontradas na face anterior da região tibial, presente em até 15% dos pacientes com doença de Crohn (DC) e 10% com retocolite ulcerativa (RCU). Apesar de até 90% dos casos de EN estarem associados à atividade da doença intestinal, sua gravidade não é necessariamente proporcional. Podem estar presentes sintomas sistêmicos como febre e artralgia por exemplo.16–18
O pioderma gangrenoso é dermatose neutrofílica muitas vezes debilitante, que cursa com úlcera dolorosa única ou múltipla de bordos vermelho‐escuro ou violáceo, irregulares, elevadas e solapadas. Varia de 2 a 20 cm e pode vir acompanhado de febre, artralgia e/ou mialgia. Tem maior prevalência em mulheres e afrodescendentes.16–18
A síndrome de Sweet, também classificada como uma dermatose neutrofílica, é mais raramente associada à DII. Geralmente cursa com febre, alterações oculares, leucocitose, pápulas, placas e nódulos eritematosos dolorosos assimétricos pelo corpo.16–18
A Síndrome de dermatose‐artrite associada a doença intestinal (BADAS) tem sua patogênese baseada no excesso de proliferação bacteriana causando liberação de antígenos e por consequência deposição de imunocomplexos. Caracteriza‐se por episódios recorrentes de febre, dor abdominal, artrite e pode apresentar‐se com máculas, pápulas, vesículas e pústulas com ou sem dor local.17
As vasculites são comuns. Geralmente se manifestam com púrpura palpável, úlceras, nódulos, vasculite digital e exantema maculopapular. Os subtipos mais descritos são vasculite leucocitoclástica, a necrosante e a linfocítica. A vasculite necrosante mais associada é a poliarterite nodosa (PAN).
Existem ainda as manifestações por má absorção, secundárias à doença gastrintestinal. É o caso da hiperceratose folicular ou frinoderma da deficiência de vitamina A, queilite angular da deficiência de vitamina B12, escorbuto pela deficiência vitamina C e acrodermatite enteropática do zinco, por exemplo.17,18
Atualmente, com o advento do uso dos imunobiológicos, alterações após o uso de terapia anti‐TNF, podendo surgir paradoxalmente lesões psoriasiformes e/ou eczematosas, além de vasculites, síndromes lúpus símile, dentre outras.16
a) Doença de Crohn
Pode‐se dividir as alterações cutâneas da DC em lesões específicas e inespecíficas. As alterações específicas são originadas por continuidade, como úlceras orais, abscessos, fissuras e fístulas perianais ou periostomais; ou pela doença de Crohn metastática (DCm), que pode produzir pápulas, placas infiltradas eritematosas, nódulos ou úlceras, à distância dos sítios de acometimento da enfermidade com ou sem dor local.16–19
Com fisiopatogenia pouco esclarecida, a DCm tem apresentação heterogênea e pode aparecer antes, durante ou após os sintomas gastrintestinais ocorrerem. Afeta ambos os sexos da mesma maneira e costuma acometer a região perineal com edema, eritema e fissuras.19
As manifestações inespecíficas podem estar relacionadas à reatividade cutânea devido aos mecanismos imunológicos da DC, serem enfermidades independentes, mas com associação à DC ou complicações secundárias às lesões causadas pela DC (tabela 4).18
Manifestações inespecíficas da doença de Crohn
| Reatividade cutânea |
| Estomatite aftosa |
| Eritema nodoso |
| Pioderma gangrenoso |
| Síndrome de Sweet |
| Sindrome de dermatose‐artrite associada a doença intestinal (BADAS) |
| Pioestomatite vegetante |
| Vasculite leucocitoclásica |
| Enfermidades independentes associadas |
| Psoríase |
| Amiloidose secundária |
| Vitiligo |
| Epidermolise bolhosa adquirida |
| Alopecia Areta (AA) |
| LES |
| Complicações secundárias |
| Acrodermatite enteropática |
| Queilite angular |
Adaptado de Gravina et al.18
b) Retocolite ulcerativa
Entre 5 a 11% dos pacientes com RCU podem apresentar alterações cutâneas. O pioderma gangrenoso e a pioestomatite vegetante são mais frequentes na RCU do que na DC. Vasculite leucocitoclástica, eritema nodoso e até mais raramente lesões orais também estão descritas.16,17
Doença celíacaA manifestação extraintestinal mais comum é a dermatite herpetiforme (DH). Causada pela deposição de imunocomplexos com IgA. A DH se expressa por pápulas e vesículas pruriginosas que se localizam preferencialmente nos cotovelos, joelhos e glúteos.20
Síndrome de Peutz‐JeghersA síndrome de Peutz‐Jeghers é doença genética caracterizada por polipose hamartomatosa intestinal e hiperpigmentação perioral típica.21 As lesões pigmentares são lentigos que podem estar presentes desde o nascimento e localizam preferencialmente ao redor dos lábios, mucosa oral, língua e nariz.22
Doença de Cronkhite‐Canada (DCC)DCC é doença rara de etiologia ainda não bem estabelecida, caracterizada por polipose adenomatosa intestinal. Na pele, é típico surgirem áreas de hipermelanose lentiginosa, alterações ungueais atróficas e alopecia. A alopecia pode apresentar características sugestivas da alopecia areata, podendo acometer também sobrancelhas, cílios, região axilar e pubiana; além disso, após períodos de disabsorção, pode estar associada a eflúvio telógeno agudo. Vitiligo, lúpus eritematoso sistêmico (LES), esclerodermia e hipotireoidismo foram descritos em pacientes com DCC.23
Doenças hepáticasExistem diversas doenças que acometem o fígado e vias biliares e também as mucosas e a pele. Serão abordadas a cirrose, a hemocromatose, as hepatites virais e também a porfiria cutânea tarda e a doença de Wilson.
a) Cirrose
A cirrose hepática pode causar diferentes sinais semiológicos. O eritema palmar, aranhas vasculares, hemangioma arteriovenoso (facial), circulação colateral em cabeça de medusa, flushing, facies pletórica, angioendoteliomatose, prurido, alterações ungueais, queda de cabelo, rarefação dos pelos corporais, vasculite, reações urticariformes e icterícia são manifestações comuns.24
As alterações ungueais são inespecíficas, podendo surgir em outras doenças como a síndrome nefrótica ou após quimioterapia por exemplo. Mesmo assim, estão classicamente associadas as unhas de Terry, apresentando‐se com o leito ungueal branco com faixa distal rosa ou acastanhada; e as unhas de Muehrcke, com faixas brancas transversais.24 Baqueteamento digital e contratura de Dupuytren também podem ser encontradas
b) Hemocromatose
Vide o tópico sobre doenças cardiovasculares acima.
Alterações secundárias à cirrose hepática também podem estar presentes.
c) Hepatite viral
Existem vários achados cutâneos relacionados às hepatites virais. Em todas elas podem ser encontradas icterícia e urticária. Na hepatite B e C, pode haver urticária‐vasculite.24
No paciente com hepatite B aguda pode ocorrer edema periorbital, exantema, reações liquenoides, doença do soro símile e PAN.24
No quadro crônico estão descritas crioglobulinemia mista, vasculite, exantema, síndrome de Gianotti‐Crosti.24,25
Líquen plano, lesões liquenoides, granuloma anular, PAN e livedo reticular foram encontrados após imunização para hepatite B.25
A hepatite C tem relação com prurido, crioglobulinemia, porfiria cutânea tarda, vasculite, livedo reticular, líquen plano, síndrome de Sjögren, urticária e PAN.24,26
Menos comumente associados estão EN, eritema multiforme, telangiectasia nevoide, pioderma gangrenoso, vitiligo, distrofia ungueal, psoríase, síndrome de Behçet, granuloma anular e poroqueratose.26
O eritema acral necrolítico que se apresenta com placas liquenificadas e bordas eritematosas podendo ter descamação, prurido e dor; é uma doença rara e está associado à hepatite C crônica.24
O hepatocarcinoma, uma das complicações da infecção crônica pelo vírus C, tem relação bem estabelecida com a crioglobulinemia mista, a PCT e com o líquen plano.24
d) Porfiria cutânea tarda
A porfiria cutânea tarda (PCT) é doença causada pela deficiência da enzima hepática uroprofirinogênio descarboxilase que altera a síntese do heme e afeta o fígado e a pele. A fotossensibilidade é a regra e manifesta‐se como vesico‐bolhas que geralmente evoluem com milia, alteração pigmentar e cicatrizes. Outros achados são hipertricose e lesões fibrosas cicatriciais.24
e) Doença de Wilson
A doença de Wilson (DW) é distúrbio do metabolismo do cobre que leva a seu acúmulo no fígado, mas também em outros órgãos e tecidos. As manifestações mucocutâneas podem ocorrer em decorrência dessa deposição, secundária ao tratamento com D‐penicilamina ou como repercussão de cirrose hepática. As alterações mais descritas são hiperpigmentação distal nos membros inferiores, a lúnula azul e o anel de Kayser‐Fieischer (círculo de pigmentação na periferia da córnea).27
A D‐penicilamina, medicamento usado no tratamento da doença de Wilson, pode alterar a biossíntese do colágeno. Com isso, pode contribuir para o surgimento de anetodermia, elastosis perforans serpiginosa, cútis laxa e pseudoxantoma elástico. Após longo período de uso, também pode predispor a reações de hipersensibilidade e doenças bolhosas.27
Foi observada maior quantidade de lipomas subcutâneos nos pacientes com DW, contudo sua fisiopatogenia ainda não foi elucidada.27 Freg e colaboradores, em 2016, descreveram caso de uma paciente de 17 anos que evoluiu com o surgimento de pioderma gangrenoso.28
Doenças oncológicasMuitas condições cutâneas podem se correlacionar a malignidades internas de formas específicas ou não específicas. Alguns critérios sugeridos por Curth podem aferir se a associação entre a lesão cutânea e a neoplasia é mais ou menos provável conforme explicitado na tabela 5.29
Postulados de Curth (Critérios de associação entre dermatose e neoplasia)
| Início simultâneo |
| Curso paralelo |
| Uniformidade de neoplasia (local ou tipo celular) |
| Associação estatística |
| Associação genética |
Adaptado de: Curth HO et al.29
1. Acantose nigricans: deve levantar suspeita de associação com neoplasia no caso de surgimento súbito em indivíduo não obeso na ausência de endocrinopatia. É mais comumente relacionada ao adenocarcinoma de estômago, embora possa se associar a outros tumores da cavidade abdominal (tratos gastrintestinal e geniturinário). Em mais de 95% dos casos, os tumores são adenocarcinomas.30
2. Síndrome de Bazex (acroceratose paraneoplásica): é erupção psoriasiforme nas superfícies acrais que pode evoluir para ceratodermia palmoplantar e distrofia ungueal.30 Orelhas, nariz, região bucinadora, mãos, pés e joelhos são frequentemente acometidos.31 Associado aos carcinomas de células escamosas do trato aerodigestivo superior (laringe, faringe, traqueia, brônquios, esôfago superior), geralmente acompanhado de metástases para linfonodos cervicais.30 Mais raramente pode ocorrer no contexto de outras neoplasias sólidas e linfomas, sendo indicada investigação da cabeça, do pescoço e da pelve.
3. Dermatoses bolhosas: há maior probabilidade de estarem relacionadas a neoplasias se a biopsia da pele revelar imunofluorescência direta negativa (ou se esta tiver depósitos de lineares de IgA na membrana basal) e se apresentar lesões mucosas exuberantes.
Pênfigo paraneoplásico: associa‐se a malignidades do sistema linforreticular (doença de Castleman, linfoma não Hodgkin, timoma, sarcoma folicular de células dendríticas, leucemia linfocítica crônica).32 Pode ter envolvimento multissistêmico com lesões brônquicas levando à falência respiratória e tem prognóstico reservado.
Variante antiepiligrina do penfigoide cicatricial secundário a adenocarcinomas.33
Dermatite herpetiforme: aumento do risco relativo de linfoma intestinal, assim como observado na própria doença celíaca.34
Epidermólise bolhosa adquirida: raramente pode ser observada em pacientes com tumores do sistema linforreticular.
Porfiria cutânea tarda: relacionado a tumores hepáticos, mas essa associação parece ser incidental uma vez que a infecção pelo vírus da hepatite C é fator de risco para ambos.
4. Dermatomiosite: 10 a 30% dos portadores de dermatomiosite podem ter neoplasias associadas (as síndromes de sobreposição e a dermatomiosite infantil raramente estão associadas a neoplasias). Quando há neoplasia associada, a doença é mais resistente ao tratamento com corticoides, sendo necessário uso de imunossupressores adjuvantes. Câncer da mama e ovário nas mulheres e câncer do pulmão e próstata no homem têm forte associação com a dermatomiosite e menor com a polimiosite. A neoplasia geralmente é encontrada no primeiro ano da doença.35
5. Ceratoses seborreicas eruptivas (sinal de Leser‐Trelat): pode ser compreendido como uma variante da acantose nigricans e, portanto, também se relacionar a adenocarcinomas do trato gastrointestinal, embora também tenha sido descrito como secundário a tumores do sistema reprodutivo feminino e desordens linfoproliferativas.36
6. Eritrodermia, prurido generalizado e ictiose: Apresenta associação predominante com malignidades linfoproliferativas, e também neoplasias sólidas. Recomenda‐se o rastreio de neoplasias em todo paciente com eritrodermia de etiologia indefinida. A pitiríase rotunda parece ser uma variante da ictiose adquirida e se correlaciona com hepatocarcinoma.
7. Eritema figurado: apenas o eritema gyratum repens parece estar associado a malignidades.37 O câncer de pulmão é a neoplasia mais comum, seguido pelos tumores da mama, trato genitourinário e gastrintestinal.
8. Hipertricose lanuginosa adquirida: é das associações cutâneas mais consistentes com neoplasias;38 o adenocarcinoma do trato gastrintestinal é o mais comum.
9. Tromboflebite migratória (síndrome de Trousseau): associação com neoplasia de pâncreas, pulmão, estômago, próstata, sistema hematopoiético.39
10. Retículo‐histiocitose multicêntrica: lesões cutâneas nodulares com predileção pelas mãos, associação com neoplasias de órgãos sólidos e linforreticulares.
11. Micose fungoide: é linfoma T cutâneo primário e pode estar associado a doença de Hodgkin e a linfoma não Hodgkin.
12. Xantogranuloma necrobiótico: associação com mieloma múltiplo.
13. Doença de Paget mamária e extramamária: possível associação com adenocarcinoma metastático da mama, neoplasias do trato gastrintestinal ou genitourinário de acordo com a área de continuidade das lesões na pele.
14. Dermatoses neutrofílicas e pioderma gangrenoso: relacionados à leucemia mieloide, principalmente na variante bolhosa do pioderma gangrenoso.
15. Paquidermoperiostose: associação com câncer de pulmão.
16. Palmas em tripas: se isolado, associa‐se ao câncer de pulmão. Se presente com acantose nigricans, investigar associação com câncer gástrico.
17. Vitiligo: pode estar associado ao melanoma, especialmente se surgir após a quarta década de vida.
Síndromes secretoras de hormônios1. Síndrome carcinoide: ver tópico sobre doenças cardiovasculares.
2. Síndrome adrenocorticotrópica ectópica: hiperpigmentação intensa e mais pronunciada do que costuma ser observada na síndrome de Cushing associada ao carcinoma de pequenas células do pulmão.
3. Síndrome glucagonoma: associada a neoplasias de pâncreas produtoras de glucagon e tem o eritema migratório necrolítico como sua manifestação cutânea.
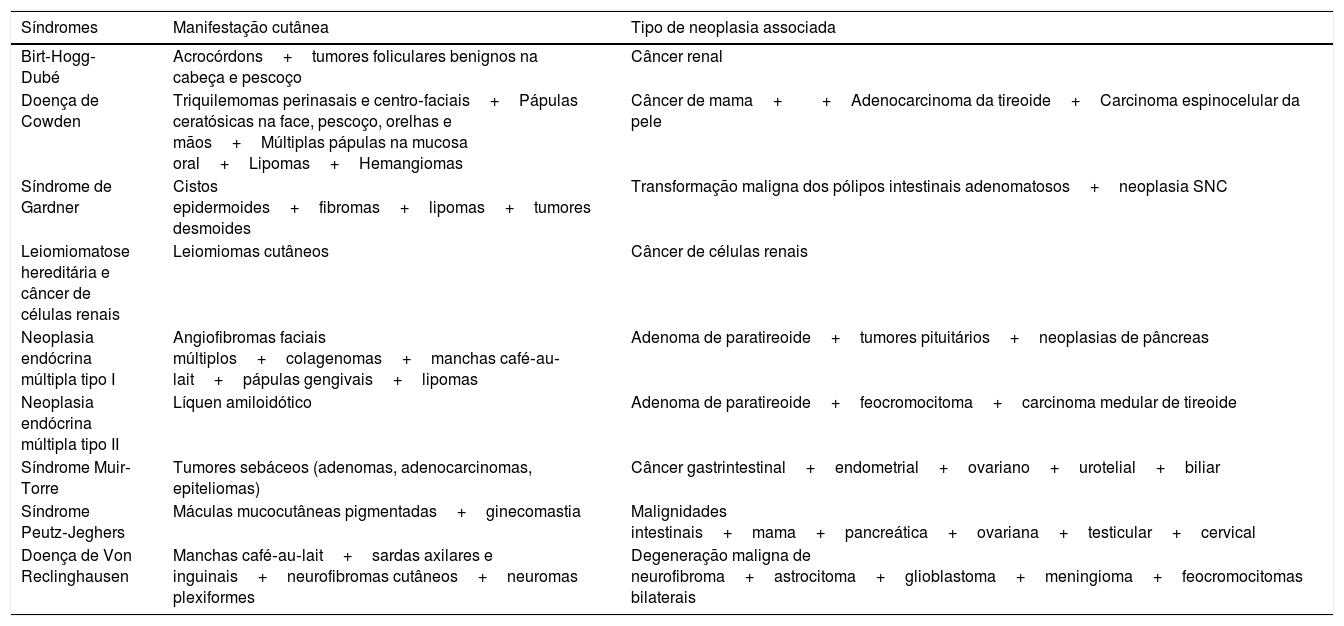
Síndromes hereditárias associadas a neoplasias internas (tabela 6)
Síndromes herdadas associadas à neoplasias internas
| Síndromes | Manifestação cutânea | Tipo de neoplasia associada |
|---|---|---|
| Birt‐Hogg‐Dubé | Acrocórdons+tumores foliculares benignos na cabeça e pescoço | Câncer renal |
| Doença de Cowden | Triquilemomas perinasais e centro‐faciais+Pápulas ceratósicas na face, pescoço, orelhas e mãos+Múltiplas pápulas na mucosa oral+Lipomas+Hemangiomas | Câncer de mama++Adenocarcinoma da tireoide+Carcinoma espinocelular da pele |
| Síndrome de Gardner | Cistos epidermoides+fibromas+lipomas+tumores desmoides | Transformação maligna dos pólipos intestinais adenomatosos+neoplasia SNC |
| Leiomiomatose hereditária e câncer de células renais | Leiomiomas cutâneos | Câncer de células renais |
| Neoplasia endócrina múltipla tipo I | Angiofibromas faciais múltiplos+colagenomas+manchas café‐au‐lait+pápulas gengivais+lipomas | Adenoma de paratireoide+tumores pituitários+neoplasias de pâncreas |
| Neoplasia endócrina múltipla tipo II | Líquen amiloidótico | Adenoma de paratireoide+feocromocitoma+carcinoma medular de tireoide |
| Síndrome Muir‐Torre | Tumores sebáceos (adenomas, adenocarcinomas, epiteliomas) | Câncer gastrintestinal+endometrial+ovariano+urotelial+biliar |
| Síndrome Peutz‐Jeghers | Máculas mucocutâneas pigmentadas+ginecomastia | Malignidades intestinais+mama+pancreática+ovariana+testicular+cervical |
| Doença de Von Reclinghausen | Manchas café‐au‐lait+sardas axilares e inguinais+neurofibromas cutâneos+neuromas plexiformes | Degeneração maligna de neurofibroma+astrocitoma+glioblastoma+meningioma+feocromocitomas bilaterais |
É definido como sensação desconfortável que desencadeia a vontade de coçar.40 Pode ser desencadeado na pele, diretamente, por estímulos mecânicos ou térmicos ou, indiretamente, por mediadores químicos ou ainda, no sistema nervoso periférico, por alteração na inervação local, ou no sistema nervoso central sem estímulo periférico. Os principais mediadores envolvidos são histamina, neuropeptídeos, prostaglandinas, serotonina, acetilcolina ou bradicinina e tantos outros estão em estudo, como os receptores vaniloides, opioides e canabinoides.
Pode ser agudo (até 6 semanas) ou crônico (mais de 6 semanas de duração). O agudo é mais relacionado à urticária, eventos adversos de fármacos e infecções e, crônico, mais associado a doenças cutâneas e sistêmicas. O prurido crônico é mais refratário ao tratamento, e, quando perpetuado no ciclo prurido‐coceira, desencadeia lesões cutâneas secundárias (escoriações, alteração da pigmentação, liquenificação) que, a longo prazo, interfere no sono, estado emocional, desejo sexual, impactando negativamente a qualidade de vida.41
A investigação do prurido se inicia por uma anamnese detalhada. Importante caracterizar o prurido (generalizado×localizado, data do início, variação ao longo do dia e fatores que intensificam ou amenizam) e questionar sobre uso de fármacos, viagens recentes, acometimento de outros membros da família, local de trabalho, contato com animais e/ou alérgenos e sinais sistêmicos, como febre, sudorese noturna, perda ponderal, dor abdominal, tonteira, artrite, parestesias. Em seguida, o exame físico, incluindo as regiões genital, interdigital, olhos e mamilos, na tentativa de identificar lesões inflamatórias ou infecciosas primárias que permitam o diagnóstico de alguma afecção dermatológica.42 Se não identificada, é necessário avaliar os remédios em uso, pois podem causar prurido seja por alergia, colestase, xerose cutânea, acúmulo de metabólitos na pele ou nervos e fototoxicidade. Se um ou mais forem suspeitos, devem ser descontinuados por 4 a 6 semanas. Havendo alívio, o nexo causal se reforça e deverá ser trocado.43 Se não houver medicamento suspeito, deve‐se considerar a possibilidade de doença neurológica ou sistêmica e exames laboratoriais devem ser solicitados: hemograma; funções renal, hepática e tireoidiana; ferro, ferritina; PCR e VHS. Se for necessário, complementa‐se com sorologia anti‐HIV, para hepatites B e C, exames de imagem do tórax e abdome, imunoeletroforese de proteínas, paratormônio, colesterol e frações e biópsia cutânea.
As causas neurológicas abrangem neuropatias (periféricas e centrais) e psicogênicas. Devemos classificar as neuropatias periféricas em: localizada (mononeuropatia) – neuralgia pós‐herpética (lesões secundárias no dermátomo afetado); notalgia parestésica (placa hiperpigmentada dorsal, na altura das vértebras T2‐T6); prurido braquiorradial (prurido e escoriação localizada na lateral dos braços e ombros) – generalizada (polineuropatia): neuropatia diabética, deficiências nutricionais, alcoolismo e doenças autoimunes. As neuropatias centrais, mais raras, abrigam os tumores do sistema nervoso central, a esclerose múltipla (xerose, lesões secundárias, com ataxia, diplopia, disestesia) e os acidentes vasculares encefálicos (lesões secundárias, com perda sensorial, fraqueza, ataxia). O prurido neuropático pode ser tratado topicamente com capsaicina e cremes à base de mentol e, por via oral, com gabapentina, pregabalina, mirtazapina e paroxetina. Já as psicogênicas são secundárias a doenças como depressão, transtorno obsessivo‐compulsivo, fibromialgia e delírio de parasitose e devem ser encaminhados para avaliação psiquiátrica. As causas psicogênicas podem ser aventadas quando não houver causa somática ou cutânea identificada, durar mais de 6 meses, houver relação temporal com eventos psicológicos, variação de intensidade com o estresse, predominar durante o repouso e aliviar com drogas psicotrópicas ou terapia.44
As causas sistêmicas incluem uma miríade de enfermidades, com suas peculiaridades. Seguem alguns exemplos:
- 1.
Hepático: cirrose biliar primária, hepatite C, colestase da gestação, induzida por fármaco. Início acral (pés e mãos), com posterior generalização. Sinais associados: icterícia, telangiectasias, edema periférico. Tratamento com colestiramina, ácido ursodesoxicólico e naltrexona.
- 2.
Renal (dialíticos, em especial): muito intenso, pior à noite, podendo evoluir para prurigo nodular, doença de Kyrle. A área mais acometida é o dorso (sinal da borboleta – área central poupada), seguida de braços, cabeça e abdome. Tratamento com fototerapia UVB, talidomida e tacrolimus tópico.
- 3.
Hematopoiético: linfoma (pode ser o primeiro sintoma da doença), mastocitose, policitemia vera (ocorre após o banho; sinais associados: equimose, sangramento de mucosa, cefaleia, hepatoesplenomegalia), anemia ferropriva (estomatite angular, palidez cutâneo‐mucosa; reverte com a reposição de ferro), leucemias, mieloma múltiplo (equimose, neuropatia, hepatoesplenomegalia), mielodisplasias. O linfoma cutâneo de células T e a síndrome de Sézary causam prurido de difícil controle.
- 4.
Neoplasias sólidas: o prurido como manifestação paraneoplásica. Precedendo ou acompanhando a neoplasia.
- 5.
Endocrinológico: no hipertireoidismo, podendo ser o primeiro sintoma da doença; pode ter tremor fino, hiperidrose, mixedema pretibial. No diabetes, prurido mais localizado, presente no tronco e couro cabeludo. Na mastocitose: refratário, com presença de urticária, angioedema, intolerância ao calor, alterações pulmonares, gastrintestinais ou cardíacas. No hipotireoidismo: menos frequente e muito relacionado à xerose; sinais associados: alopecia, edema periorbital, macroglossia, aumento de peso, fadiga.
- 6.
Colagenoses: avaliar os medicamentos usados, pois podem causar direta ou indiretamente prurido. Na dermatomiosite, síndrome de Sjögren e lúpus, o prurido pode preceder a doença em até 10 anos. Já na esclerodermia, o prurido é tardio. Dermatomiosite: importante sintoma da doença; avaliar uso de fotoproteção e avaliar efeito colateral de medicamentos, como o antimalárico que causa farmacodermia em até 30% dos casos. Esclerodermia: o prurido ocorre mais à noite e nas áreas afetadas, sendo agravado por estresse, fadiga, sono, água e ambientes quentes, com dormência e parestesia associadas. Para tratamento, otimizar hidratação cutânea e avaliar medicamentos usados (bloqueadores de canal de cálcio causam prurido e pode ser trocado por inibidores da receptação da serotonina), e investigação de neoplasias. Síndrome de Sjögren: o olho seco pode ser pruriginoso, e a xerose cutânea agravar a sintomatologia. Se não há controle com a hidratação cutânea, deve‐se procurar por complicações da doença, como vasculite, neuropatia, cirrose biliar primária, hepatite autoimune. Lúpus eritematoso: o prurido pode ocorrer mesmo na ausência de lesão. Avaliar fotoproteção, otimizar tratamento (cessar tabagismo), avaliar efeitos adversos (talidomida causa neuropatia, antimalárico causa prurido) e complicações (nefrite, cirrose biliar primaria, hepatite autoimune e colangite esclerosante).45
- 7.
HIV: o prurido pode estar associado a dermatoses que são mais prevalentes ou que são agravadas pelo HIV, ou ocorrer como sintoma primário da infecção, como na foliculite eosinofílica, reação exacerbada à picada de inseto e pápulas pruriginosas do HIV.
Prurido é sintoma comum e incômodo. Seu diagnóstico pode ser fácil e objetivo, mas pode também ser desafiador. Seu estudo está em progresso, e novos fármacos estão em desenvolvimento.
Manifestações cutâneas associadas à COVID‐19.O comprometimento da pele parece ser um efeito indireto e não específico da COVID‐19, assim como nas demais infecções virais, independente do estágio ou gravidade da doença. No entanto, há estudos tentando comprovar a presença do vírus diretamente na pele, causando lesão cutânea. Estas últimas seriam classificadas como lesões específicas da COVID‐19.
Um número consistente de relatos de casos e séries clínicas de diversas regiões do mundo foi publicado, descrevendo um espectro de manifestações cutâneas associadas à infecção por SARS‐CoV‐2. No entanto, imagens e achados histopatológicos dessas lesões raramente foram incluídos, dificultando a correlação causal entre a manifestação clínica dermatológica e a COVID‐19. Não há um consenso; no entanto, o grupo italiano conduzido porPaolo Gisondi sugeriu que as manifestações cutâneas fossem classificadas em quatro categorias.46
Categorias das manifestações cutâneas da COVID‐19Exantema (erupção cutânea tipo varicela, papulo‐vesicular e morbiliforme)Um estudo conduzido por Galván Casas et al., na Espanha, mostrou que erupções vesiculares geralmente surgem no início da doença, antes mesmo de outros sintomas. Já o padrão chilblain‐like costuma surgir tardiamente, enquanto as outras manifestações costumam acompanhar os demais sinais e sintomas da infecção.47
A erupção vesicular descrita na COVID‐19 costuma se manifestar na fase inicial da doença e clinicamente se caracteriza por lesões papulo‐vesiculares monomórficas (diferenciando‐a da varicela), geralmente generalizadas, com acometimento predominante do tronco, com ou sem prurido.48 As lesões surgem em média três dias após o início dos sintomas, com duração aproximada de oito dias. Relacionada à doença de gravidade moderada, e geralmente acomete pacientes de meia‐idade.46
O exantema maculopapular ou morbiliforme se assemelha clinicamente aos demais exantemas virais. Pode apresentar uma distribuição perifolicular, acometendo dobras axilares, fossa cubital ou ser ocasionalmente disseminado. As lesões costumam acompanhar os demais sintomas da infecção viral e têm duração de 3 a 10 dias. O prurido é descrito na maioria dos pacientes e geralmente está associada a doença mais grave e em pacientes mais idosos.46,47
Manifestação vascular (lesões chilblain‐like, petequiais/purpúricas e livedoides)Diversos tipos de lesões vasculares foram descritos na infecção por SARS‐CoV‐2, incluindo lesões chilblain‐like, livedoides, petequiais, purpúricas e necróticas. A maioria parece ter em sua fisiopatologia a formação de trombos intravasculares. Casos de púrpura trombocitopênica imune (PTI) e síndrome do anticorpo antifosfolipídio (SAF) foram descritos como manifestações cutâneas associadas à COVID‐19. O espectro das lesões vasculares pode ter diferentes mecanismos de ação, incluindo a ação direta do vírus nas células endoteliais e/ou efeito indireto desencadeando reações imunes ou autoimunes (como no caso da PTI). Seja qual for o mecanismo, a consequente disfunção microvascular pode levar ao aumento da vasoconstrição, inflamação e um estado de pró‐trombótico.46
Lesões chilblain‐like ocorrem normalmente em crianças e adolescentes e afetam principalmente as regiões acrais. Manifestam‐se clinicamente como máculas eritemato‐violáceas, que podem evoluir com vesícula, pústulas ou bolhas.49 Geralmente são assimétricas e apresentam em sua maioria dor o e/ou prurido. Afetam principalmente pacientes jovens na ausência de sintomas sistêmicos. As lesões costumam surgir na fase mais tardia da infecção e parecem ter resolução espontânea após 2 a 4 semanas.46 O surgimento tardio das lesões pode explicar os possíveis resultados de detecção viral negativos.47
Um amplo espectro de lesões purpúricas e petequiais foi descrito como possível associação à infecção por SARS‐CoV‐2. As lesões podem surgir em qualquer momento durante o curso da doença e sem predomínio de localização. Podem se apresentar apenas como petéquias ou surgir como máculas eritemato‐purpúricas que coalescem formando placas, púrpura retiforme e até mesmo lesões necróticas. As lesões necróticas podem surgir em qualquer momento durante o curso da doença e se localizam preferencialmente nos membros inferiores e em pacientes mais idosos ou na forma grave da doença. Há relatos de lesões semelhantes ao livedo reticular e acrocianose em graus variáveis, de acordo com os autores, as lesões podem ser explicadas pelos distúrbios de coagulação subjacentes associados com COVID‐19.46,47
Foram relatados casos de crianças com sinais e sintomas consistentes com doença de Kawasaki e evidência laboratorial de infecção recente por SARS‐CoV‐2. Alguns casos foram denominados provisoriamente como síndrome inflamatória multissistêmica pediátrica associada à infecção por SARS‐CoV‐2. Tal associação pode corroborar a ocorrência de um dano endotelial associado à infecção pelo vírus.46,50
Erupção acropapularesA erupção papular acral consiste no surgimento de pápulas pruriginosas eritemato/amareladas confluentes que, após alguns dias, formam placas pruriginosas, simétricas, refratárias ao tratamento com esteroides tópicos. As lesões surgiram cerca de 13 dias após a positividade para o SARS‐CoV‐2, com resolução espontânea.51
Erupção urticariformeO padrão urticariforme foi descrito como uma erupção cutânea eritematosa levemente pruriginosa com resolução espontânea em poucos dias que costuma surgir concomitante com os demais sintomas da COVID‐19.47
A ativação de mastócitos e basófilos, por efeito direto ou indireto viral é um evento possível e, portanto, manifestações cutâneas como urticária e exacerbação de condições preexistentes como dermatite atópica podem ser sinais clínicos iniciais da COVID‐19. O efeito citopático direto do SARS‐CoV‐2 pode ocorrer em lesões vesiculares ou papulovesiculares, que são muito semelhantes aos causados pela família Herpesviridae.
Em conclusão, as manifestações cutâneas associadas à infecção pelo SARS‐CoV‐2 podem ser múltiplas e de etiologia específica, de difícil comprovação etiológica ou, mais frequentemente, serem para‐virais, em decorrência de fenômenos inflamatórios liberados no curso da infecção ou, ainda, pela exposição a fármacos durante o período prodrômico ou de tratamento da doença.52
Suporte financeiroNenhum.
Contribuição dos autoresJuliana Martins Leal: Elaboração e redação do manuscrito; aprovação da versão final do manuscrito.
Gabriela Hygino Souza: Elaboração e redação do manuscrito; aprovação da versão final do manuscrito.
Paula Figueiredo de Marsillac: Elaboração e redação do manuscrito; aprovação da versão final do manuscrito.
Alexandre Carlos Gripp: Elaboração e redação do manuscrito; aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Leal JM, Souza GH, Marsillac PF, Gripp AC. Skin manifestations associated with systemic diseases – Part II. An Bras Dermatol. 2021;96:672–87.
Trabalho realizado no Hospital Universitário Pedro Ernesto, Rio de Janeiro, RJ, Brasil.
