












A pele demonstra o que acontece no organismo em muitas doenças porque reflete na superfície cutânea alguns processos internos. Nesse sentido, ela, como órgão, extrapola as suas funções de proteção e barreira, pois dá pistas para o reconhecimento de algumas enfermidades sistêmicas. O dermatologista, então, levanta hipóteses diagnósticas de afecções relacionadas a todos os sistemas e direciona para a especialidade adequada. Aliado ao olhar treinado, por seu fácil acesso, a pele ainda é o local ideal para realização de biópsias, nas quais a histopatologia pode ser específica ou não específica da doença de base; no primeiro caso, essa avaliação sela o diagnóstico da enfermidade em si. As manifestações não específicas podem ocorrer em vários contextos e nesse caso a histopatologia não é própria de uma determinada doença. Este artigo é dividido em duas partes que abordarão grandes grupos de doenças. Nesta primeira parte, descrevem‐se as manifestações cutâneas das principais doenças reumatológicas, que são as de maior interface com a dermatologia. Falamos também das manifestações vasculares e das doenças granulomatosas. Na segunda parte, abordaremos as doenças endocrinológicas, hematológicas, oncológicas cardiovasculares, renais, gastrointestinais, o prurido e suas causas e, por último, as manifestações dermatológicas da infecção pelo coronavírus SARS‐CoV‐2. Nossa pretensão é, usando uma linguagem direta e de fácil acesso, conseguir disponibilizar um material prático para servir de consulta e aprimoramento a todos os dermatologistas que reconhecem a importância do olhar integral sobre o paciente.
A pele se comunica com o organismo, muitas vezes deixando transparecer os primeiros sinais e/ou sintomas de uma doença interna – podendo, inclusive, ser essas as únicas expressões de doenças sistêmicas. Nesta primeira parte de nosso trabalho, abordamos de maneira abrangente as manifestações cutâneas das colagenoses, as doenças neutrofílicas, as púrpuras e vasculites e as doenças granulomatosas. Em algumas dessas doenças é possível separar as manifestações cutâneas em específicas (ao serem biopsiadas, a histopatologia representa e sela o diagnóstico da doença investigada) e inespecíficas da doença (não há características histopatológicas da doença). Em outras, essa classificação não é aplicável. Descreveremos essa divisão, quando possível.
Lúpus eritematoso sistêmicoO lúpus eritematoso (LE) é doença inflamatória do tecido conjuntivo com diversidade de apresentações clínicas, caracterizada pela produção de autoanticorpos contra constituintes celulares. Os critérios EULAR/ACR de 2019 para lúpus eritematoso sistêmico (LES) incluem FAN positivo pelo menos uma vez como critério obrigatório, seguido por critérios adicionais separados em sete grupos clínicos (constitucionais, hematológicos, neuropsiquiátricos, mucocutâneo, serosas, musculoesquelético e renal) e três imunológicos (anticorpos antifosfolipídios, proteínas do complemento e anticorpos específicos para o LES).1 Na atual classificação, as lesões cutâneas são divididas em quatro subtipos, com pontuação distinta entre elas de acordo com o risco de estarem presentes na vigência do lúpus sistêmico (alopecia não cicatricial – 2 pontos; úlceras orais – 2 pontos; lúpus cutâneo crônico discoide ou subagudo – 4 pontos; lúpus cutâneo agudo – 6 pontos). Para o diagnóstico de LES, o paciente deve apresentar no mínimo 10 pontos. As manifestações cutâneas do lúpus compõem um espectro que varia de lesões de caráter benigno e autolimitado a erupções graves, podendo, por vezes, ser fatais quando associadas ao LES.
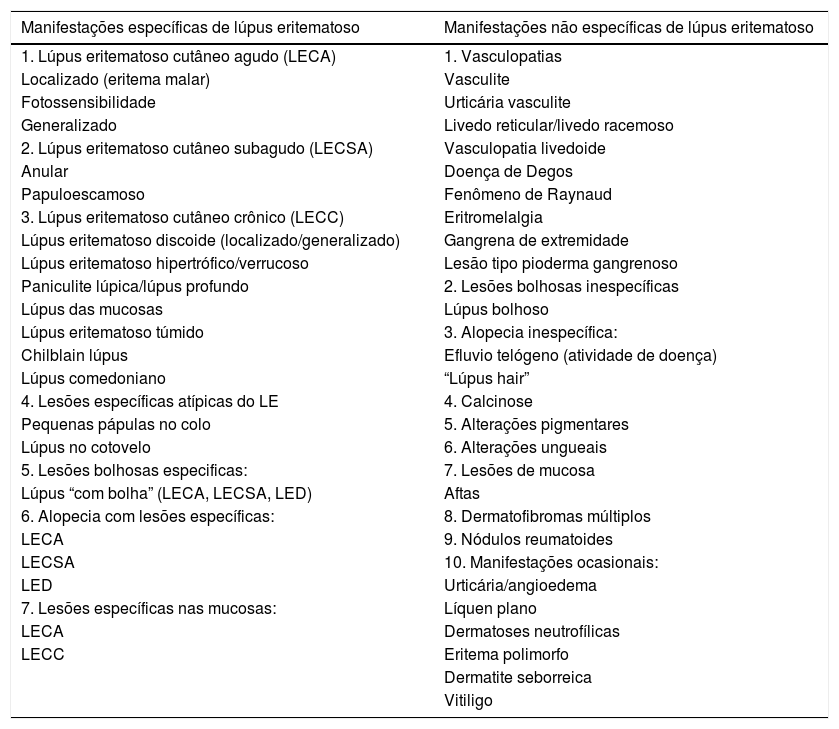
O LE pode ocorrer como manifestação do LES ou independentemente dele. Gilliam e Sontheimer classificaram o LE cutâneo em três formas, com base estritamente na aparência clínica da lesão cutânea: cutâneo crônico, cutâneo subagudo e cutâneo agudo. Gilliam, entretanto, ampliou essa classificação com base em características clínicas e histopatológicas específicas e inespecíficas.2 O paciente com LE pode apresentar mais de uma forma clínica da doença. As manifestações inespecíficas são as que não apresentam o quadro histopatológico característico do LE, mas dão “dicas” quanto ao diagnóstico e ao prognóstico. Em contraste, as manifestações específicas de LE englobam os subtipos de lúpus eritematoso cutâneo (LEC), que são o lúpus eritematoso cutâneo crônico (LECC), subagudo (LECSA) e agudo (LECA). As manifestações cutâneas do LES estão descritas na tabela 1.
Lesões específicas e não especificas do lúpus eritematoso
| Manifestações específicas de lúpus eritematoso | Manifestações não específicas de lúpus eritematoso |
|---|---|
| 1. Lúpus eritematoso cutâneo agudo (LECA) | 1. Vasculopatias |
| Localizado (eritema malar) | Vasculite |
| Fotossensibilidade | Urticária vasculite |
| Generalizado | Livedo reticular/livedo racemoso |
| 2. Lúpus eritematoso cutâneo subagudo (LECSA) | Vasculopatia livedoide |
| Anular | Doença de Degos |
| Papuloescamoso | Fenômeno de Raynaud |
| 3. Lúpus eritematoso cutâneo crônico (LECC) | Eritromelalgia |
| Lúpus eritematoso discoide (localizado/generalizado) | Gangrena de extremidade |
| Lúpus eritematoso hipertrófico/verrucoso | Lesão tipo pioderma gangrenoso |
| Paniculite lúpica/lúpus profundo | 2. Lesões bolhosas inespecíficas |
| Lúpus das mucosas | Lúpus bolhoso |
| Lúpus eritematoso túmido | 3. Alopecia inespecífica: |
| Chilblain lúpus | Efluvio telógeno (atividade de doença) |
| Lúpus comedoniano | “Lúpus hair” |
| 4. Lesões específicas atípicas do LE | 4. Calcinose |
| Pequenas pápulas no colo | 5. Alterações pigmentares |
| Lúpus no cotovelo | 6. Alterações ungueais |
| 5. Lesões bolhosas especificas: | 7. Lesões de mucosa |
| Lúpus “com bolha” (LECA, LECSA, LED) | Aftas |
| 6. Alopecia com lesões específicas: | 8. Dermatofibromas múltiplos |
| LECA | 9. Nódulos reumatoides |
| LECSA | 10. Manifestações ocasionais: |
| LED | Urticária/angioedema |
| 7. Lesões específicas nas mucosas: | Líquen plano |
| LECA | Dermatoses neutrofílicas |
| LECC | Eritema polimorfo |
| Dermatite seborreica | |
| Vitiligo |
LECA localizado: o eritema facial comumente denominado de “rash malar” é caracterizado por eritema malar e nasal e edema que poupam o sulco nasolabial. O eritema é induzido pela luz ultravioleta, e pode durar horas, dias ou semanas, involuir espontaneamente e recorrer. Nos casos de intensa fotoexposição, a superfície da pele pode demonstrar necrose – o chamado “lúpus com bolha” – e, se generalizado, assemelhar‐se à necrólise epidérmica tóxica (NET), denominado então lúpus NET‐símile (fig. 1). Fototipos elevados podem se apresentar clinicamente com hiperpigmentação ou hipopigmentação, mesmo após a resolução da inflamação. Comumente se associa ao LES com anticorpo anti‐Ro (SSA) positivo. A fotossensibilidade é um fator importante em todos os tipos de pele no LE. A radiação UVB e UVA podem agravar a doença cutânea.
.")
LECA generalizado: eritema maculopapular envolvendo áreas fotoexpostas (mãos e braços) nas regiões interarticulares. A pele que recobre as articulações dos dedos geralmente é poupada, oposto ao que ocorre no sinal de Gottron da dermatomiosite, no qual o eritema é justa‐articular.3
Histopatologia: dermatite de interface vacuolar, queratinócitos apoptóticos, infiltrado linfo‐histiocitário na derme superficial e deposição de mucina na derme.4
Lúpus eritematoso cutâneo subagudo (LECSA)Manifesta‐se inicialmente com pequenas pápulas eritematosas e ligeiramente descamativas que podem evoluir para placas psoriasiformes (forma papuloescamosa) ou eritematosas anulares com clareamento central e descamação periférica (forma anular). Os dois tipos de lesão podem ter aspecto superposto e apresentar vesículas e crostas ou até bolhas hemorrágicas na periferia, em decorrência do dano à epiderme e à camada basal pela fotoexposição – o chamado “lúpus com bolha”.3 As áreas fotoexpostas são mais comumente afetadas, como a superfície extensora dos membros superiores, pescoço e tronco superior (conhecido como “V do decote”). Ao regredirem, as lesões deixam telangiectasias, hiper ou hipocromia residuais. A perda do pigmento pode ser tão intensa a ponto de assumir características vitiligoides (fig. 2). Cicatrizes e atrofia não são observadas. O LECSA parece estar associado ao LES em aproximadamente 50% dos casos; sua histopatologia exibe vacuolização da camada basal, infiltrado linfocitário perivascular, depósito de mucina na derme e, quando comparado ao LECC, há menos hiperceratose e rolhas córneas foliculares.4
Lúpus eritematoso cutâneo crônico (LECC)
LE discoide clássico: são as lesões de LECC caracterizadas por eritema, telangiectasias, escamas lamelares aderentes, de aspecto discoide, que variam de placas finas a espessas, contendo hipo, hiper ou acromia que, quando não tratadas eficazmente, deixam atrofia e cicatrizes. Em geral, são assintomáticas, únicas ou múltiplas, localizadas preferencialmente nas áreas fotoexpostas, principalmente na face e conchas auriculares. Quanto mais extenso e abaixo do pescoço (LECC discoide disseminado), maior o risco de desenvolvimento de LES. Estima‐se que 5 a 30% dos pacientes com LECC desenvolvem a doença sistêmica. Lesões acneiformes, incluindo comedões e cicatrizes puntiformes, são apresentações atípicas denominadas lúpus comedoniano.5 Envolvimento do couro cabeludo ocorre em aproximadamente 60% dos pacientes com LED, e pode ser a única região envolvida em aproximadamente 10% dos casos. Alopecia cicatricial ocorre secundária à destruição folicular, caso não haja tratamento apropriado a tempo.
LE hipertrófico ou verrucoso: variante rara de LECC, caracterizado por placas verrucosas cuja escama é substituída por hiperceratose. As lesões localizam‐se, preferencialmente, nas superfícies extensoras dos membros, mas também no dorso e na face. Em geral, são pruriginosas. Pode haver superposição com o líquen plano (LP), chamado de LE/LP. Há poucos sintomas sistêmicos e alterações laboratoriais. As lesões são de difícil tratamento com a terapêutica convencional, mas podem responder aos retinoides orais.6
Paniculite lúpica: manifesta‐se clinicamente com nódulos subcutâneos pétreos que, se não forem devidamente tratados, levam a atrofia e calcificação subcutânea e ocasional ulceração. Ocorre nas regiões malares e temporais, nos braços, coxas, glúteos e mamas. Na histopatologia, observa‐se uma paniculite lobular com infiltração linfocítica que pode, eventualmente, organizar‐se sob a forma de folículos linfoides com centros germinativos e atingir os septos. Pode haver vasculite linfocítica e infiltrado contendo eosinófilos. O termo lúpus profundo (de Kaposi‐Irgang) muitas vezes é utilizado como sinônimo de paniculite lúpica; no entanto, alguns autores reservam esse termo para quadros de paniculite lúpica com lesões de lúpus discoide na superfície.
Lúpus túmido: é caracterizado por placas ou pápulas eritemato‐violáceas infiltradas, sem descamação, que acometem principalmente áreas fotoexpostas como a face e o pescoço. As lesões tendem a involuir sem cicatrizes ou atrofia. Pacientes com LE túmido raramente atendem aos critérios para a forma sistêmica da doença.
Perniose lúpica (chilblain lúpus): variedade rara de LECC, mais comum em locais de clima frio, localizando‐se nas extremidades ou na ponta nasal. Histopatologia: hiperceratose, rolhas córneas foliculares, alteração vacuolar da camada basal com espessamento da membrana basal, infiltrado linfocítico próximo à junção dermo‐epidérmica e presença de mucina na derme.7
Lesões específicas atípicas do LELúpus papuloso (do colo): pequenas lesões papulosas eritematosas encontradas especialmente no colo de mulheres jovens, É clinicamente distinta da mucinose cutânea associada ao LE.7
Lúpus no cotovelo (“prurigo do lúpus”): consiste em pápulas ou placas eritematosas nos cotovelos, geralmente bilaterais e recorrentes, com histopatologia específica para LE. Essa localização peculiar parece estar associada a um maior risco de envolvimento sistêmico.8
Lesões não específicas de LEVasculite: a vasculite leucocitoclástica é a manifestação de lesão inespecífica mais frequente de LES (20 a 40%). As lesões ocorrem, preferencialmente, nas extremidades dos membros inferiores ou em áreas de pressão ou traumatismo, e podem manifestar‐se como uma vasculite cutânea de pequenos vasos, com máculas, pápulas ou lesões purpúricas palpáveis. Estão relacionadas à atividade de doença.
Vasculopatias:
- a)
Livedo reticular ou livedo racemoso: geralmente encontrado em pacientes com síndrome do anticorpo antifosfolipídio (SAF). No entanto, muitos desses pacientes também apresentam LE associado com anticorpo anticardiolipina e/ou anticoagulante lúpico positivos;
- b)
Vasculopatia livedoide e atrofia alba de Millian: considerada uma vasculopatia trombótica localizada que, ao cicatrizar, deixa área de atrofia branca que costuma ser dolorosa;
- c)
Doença de Degos‐símile: pequenas lesões papulosas (2‐5mm) eritematosas com depressão central, que involuem deixando cicatriz atrófica branca com telangiectasias na periferia. Diferencia‐se de doença de Degos verdadeira por ter associação com LE e curso mais benigno e autolimitado;
- d)
Fenômeno de Raynaud: encontrado em 10 a 44% (∼33%) dos casos de LE. Associação com presença do anticorpo anti‐U1RNP (60 a 90% dos casos);
- e)
Eritromelalgia: eritema, calor, dor intolerável nas mãos e nos pés, predominantemente. Piora com exposição ao calor e alivia com o frio;
- f)
Gangrena de extremidade: fenômeno trombótico. Deve‐se pesquisar anticorpo antifosfolipídio, crioglobulinemia e endocardite infecciosa.
Lesões bolhosas: consistem nas lesões de lúpus bolhoso, cuja histopatologia é semelhante a da dermatite herpetiforme ou a da epidermólise bolhosa adquirida. As bolhas têm início agudo, predileção pelo tronco, região cervical e extremidades proximais. Apresenta cinco critérios diagnósticos propostos pelo American College of Rheumatology (ACR): 1) erupção bolhosa adquirida, não cicatricial, que surge em área fotoexposta, porém não limitada a ela; 2) histopatologia com bolha subepidérmica com infiltrado predominantemente neutrofílico na derme e na zona da membrana basal; 3) imunofluorescência direta demonstrando depósitos de IgG, IgA, IgM e C3 na zona da membrana basal da pele perilesional; 4) anticorpos circulantes contra colágeno tipo VII confirmados por imunofluorescência indireta na técnica de salt‐split skin, immunoblotting ou imunoprecipitação; 5) depósitos de imunoglobulina nas fibrilas de ancoragem e no colágeno tipo VII na microscopia imunoeletrônica.6–11
Alopecia:- a)
Específica: lesões de LECA e LECSA no couro cabeludo podem gerar alopecia não cicatricial. Já a lesão de LECC, se não tratada a tempo, pode evoluir para lesão cicatricial;
- b)
Inespecíficas: eflúvio telógeno e eflúvio anágeno: geralmente indicativos de atividade de doença (o anágeno pode ocorrer rapidamente após o início de grande reativação do LES ou após início de tratamento com ciclofosfamida ou metotrexato). Lupus hair: hastes capilares de aspecto ressecado e curtas na linha de implantação frontal. Acomete mulheres com LES em atividade de longa data.
Mucinose papulonodular: indicativa de atividade de doença e forte relação com acometimento renal. Causadas por depósito de mucina na derme, em área com ou sem alterações de LE, localizadas na região superior do tronco e nas extremidades.
Calcinose: nódulos duros e irregulares no subcutâneo que podem eliminar para a superfície, com saída de material branco a amarelado.
Alterações pigmentares: são comuns hipo e hiperpigmentação. Atenção para hiperpigmentação causada pelo antimalárico: barba, cabelos e cílios acinzentados, hiperpigmentação difusa e das bordas ungueais.
Alterações ungueais: estilhas hemorrágicas, pitting, leuconiquia, onicólise (mais comum), baqueteamento digital e lúnula vermelha, telangiectasias nas cutículas e discromias.
Lesões nas mucosas: qualquer mucosa pode ser afetada no LE. Ocasionalmente, as lesões podem apresentar histopatologia específica (de LECC ou LECA); ulcerações aftoides e erosões são observadas em 5 a 10% dos pacientes com LES.
Dermatofibromas eruptivos: o surgimento de múltiplos dermatofibromas pode acompanhar o LES e outras doenças do colágeno, como a síndrome de Sjögren.
Nódulos reumatoides: presentes em 5 a 10% dos pacientes com LES, acometem principalmente as mãos e os cotovelos.
Manifestações ocasionais: urticária/angioedema, líquen plano, eritema elevatum diutinum, pioderma gangrenoso, síndrome de Sweet, pustulose amicrobiana das dobras, eritema polimorfo, dermatite seborreica extensa e refratária, vitiligo, dermatite factícia e exantema.
DermatomiositeDermatomiosite é doença inflamatória idiopática, clinicamente heterogênea, que pode ser de difícil diagnóstico. Abrange manifestações cutâneas variadas paralelas ou não à miosite e ao envolvimento sistêmico no decorrer do tempo e/ou de gravidade.
Bohan e Peter, em 1975, sugeriram o uso de cinco critérios para diagnosticar a dermatomiosite: 1) fraqueza muscular proximal simétrica que progride ao longo de um período de semanas a meses; 2) níveis séricos elevados de enzimas musculares; 3) eletroneuromiografia anormal; 4) biópsia muscular anormal; e 5) presença de manifestações cutâneas típicas de dermatomiosite. Novos critérios, descritos por Dalakas e Hohlfeld, em 2003, destacam a importância da avaliação histológica do músculo, melhorando a especificidade, mas tornando‐se menos sensível. São critérios úteis para avaliação, porém desnecessários em paciente com doença cutânea característica, particularmente aqueles que apresentam fraqueza muscular proximal e enzimas musculares elevadas.
O curso clínico das lesões cutâneas da dermatomiosite não é necessariamente paralelo ao da doença muscular, e pode preceder ou seguir a miosite. Em mais da metade dos pacientes, as manifestações cutâneas precedem o envolvimento muscular em meses ou anos. Além disso, a atividade da doença muscular não é refletida pela atividade da doença cutânea. O envolvimento cutâneo pode ser dividido em sete tipos: patognomônico, característico, compatível, menos comum, raro, descrito recentemente e manifestações cutâneas não específicas.9 As manifestações cutâneas patognomônicas (heliotropo e pápulas de Gottron) e as demais serão relatadas a seguir (fig. 3).
. À direita: eritema no dorso das mãos (sinal de Gottron).")
Pápulas de Gottron: consistem em pápulas eritemato‐violáceas cobrindo as articulações interfalangeanas e metacarpofalangeanas.
Heliotropo: eritema rosa a violáceo, com ou sem edema, envolvendo a pele periorbitária. A pálpebra superior é a mais acometida.
Sinal de Gottron: máculas eritemato‐violáceas sobre outras articulações, como cotovelos ou joelhos.
Fotossensibilidade: o quadro clínico é marcado por intensa fotossensibilidade, com lesões eritemato‐violáceas no couro cabeludo, pescoço, ombros, superfícies extensoras das extremidades superiores, parte superior do tórax (“sinal do decote em V”) e parte superior das costas (“sinal do xale”). O eritema malar atinge o sulco nasolabial em oposição ao eritema malar no LE, que o poupa.
Prurido: pode ser intenso e persistente, afetando a qualidade de vida dos pacientes. Além disso, sua presença pode ser útil para distinguir clinicamente a dermatomiosite dos quadros de LE, onde não há prurido marcante. Muitas vezes, é resistente ao tratamento com anti‐histamínicos e corticosteroides tópicos. Lesões eritemato‐escamosas nas laterais das coxas também podem estar presentes (“sinal do coldre”).
Lesões no couro cabeludo: o envolvimento do couro cabeludo é caracterizado por placas atróficas, eritematosas e escamosas. O prurido nessa região é marcante, maior do que se pode apreciar a partir dos achados clínicos. Essas manifestações da dermatomiosite podem ser diagnosticadas erroneamente, principalmente na fase inicial, como dermatite seborreica ou psoríase. Pode ocorrer alopecia leve a moderada, não cicatrical.
Lesões nas dobras ungueais: telangiectasia periungueal (aparentes ou por microscopia digital), hipertrofia e distrofia cuticular e pequenos infartos hemorrágicos são alterações típicas das pregas ungueais.
Vasculite: pode se manifestar como púrpura palpável, lesões semelhantes à urticária, livedo reticular, infarto periungueal, ulceração digital ou oral; menos comumente, como lesões vesico‐bolhosas, erosivas, necróticas e ulcerativas. As manifestações de vasculite cutânea ocorrem principalmente na dermatomiosite juvenil, e geralmente como vasculite leucocitoclástica. A presença de lesões cutâneas vesico‐bolhosas e erosivas ou vasculite cutânea deve levar a uma investigação diagnóstica para possível malignidade subjacente.
Calcinose cutânea: depósitos calcificados na pele e tecido subcutâneo. De acordo com os níveis séricos de cálcio/fosfato, a calcinose cutânea pode ser classificada em quatro subtipos: distrófica, metastática, iatrogênica e idiopática. A calcinose distrófica ocorre em tecidos lesados e está associada a várias doenças do tecido conjuntivo, principalmente a dermatomiosite; nela, os níveis séricos de cálcio/fosfato estão dentro da normalidade. Sua fisiopatologia permanece obscura. A calcinose é mais comum na dermatomiosite juvenil (30 a 70% dos casos) do que no adulto (10% dos casos). Apresenta‐se como nódulos superficiais ou subcutâneos, encontrados principalmente em locais de trauma repetido, como glúteos, cotovelos e joelhos. A calcinose localizada na face extensora das extremidades pode ulcerar, levando a úlceras crônicas de difícil cicatrização.
“Mãos de mecânico”: caracterizada por hiperceratose, descamação e fissuras nos dedos ou mesmo nas palma das mãos. As lesões são distribuídas ao longo da face ulnar do polegar e aspecto radial dos dedos e são mais proeminentes no dedo indicador e médio, com extensão rara para a palma. É característico da síndrome anti‐sintetase, que compreende também os achados de artrite, doença pulmonar intersticial e fenômeno de Raynaud. As “mãos de mecânico” também podem ser vistas fora do contexto anti‐sintetase, em casos de polimiosite, dermatomiosite clássica e amiopática.
Variante Wong: caracterizada por pápulas eritematosas foliculares e hiperceratóticas no lado extensor das extremidades, algumas vezes associadas à ceratodermia palmar.
Eritema flagelado: caracterizada por lesões eritematosas, edematosas, lineares, pruriginosas e/ou dolorosas, como se tivessem sido produzidas por açoitamento, localizadas no tronco. Pode ocorrer em associação à dermatomiosite, doença de Still do adulto, tratamento com bleomicina sistêmica, consumo de shiitake e exposição à medusa.
Paniculite: ocorre na forma juvenil e adulta, como nódulos endurecidos, isolados ou confluentes nos glúteos, braços, coxas e abdome. Algumas lesões são seguidas de calcificação. O exame histológico mostra uma paniculite lobular com infiltrado linfoplasmocitário, liquefação na junção dermoepidérmica e alterações membranocísticas. A patogênese da paniculite na dermatomiosite não é clara.
Erupções vesicobolhosas: vesículas ou bolhas podem se desenvolver, raramente, nas superfícies dorsais das mãos ou antebraços, e em outras áreas. Essas erupções podem ser causadas por fotossensibilidade, com achados histopatológicos de degeneração por liquefação da camada basal da epiderme, edema subepidérmico, deposição de mucina na derme superior ou complicações de doenças bolhosas autoimunes.
Eritrodermia: é rara e tem associação com malignidade.
Lesões na mucosa oral: o envolvimento da mucosa oral inclui telangiectasias, edema, erosões, úlceras e áreas semelhantes à leucoplasia. A telangiectasia gengival provavelmente representa um achado diagnóstico análogo à telangiectasia da prega ungueal. A hipossalivação é comum e pode explicar um aumento da prevalência de cárie dentária nesses pacientes.
Pápulas de Gottron inversas: uma manifestação muito rara da dermatomiosite. Localizadas na superfície palmar das articulações interfalangeanas (ao contrário das pápulas de Gottron clássicas). Apresentam‐se como hiperceratose triangular branca localizada.
Manifestações cutâneas recentemente descritas: são raras e incluem as pápulas de Gottron inversas, as ulcerações digitais, as pápulas (ou sinal) de Gottron com ulceração e os pés de alpinista (pés de mecânico).
Fenômeno de Raynaud: visto principalmente em pacientes com dermatomiosite e doenças autoimunes de sobreposição ou na síndrome anti‐sintetase. É uma manifestação inespecífica da doença.9
A biópsia de pele pode ajudar a diferenciar a dermatomiosite de outras doenças papulo‐escamativas ou eczematosas, mas não pode ser usada para distinguir dermatomiosite do LE. Classicamente, a biópsia de pele na dermatomiosite demonstra hiperceratose, atrofia epidérmica, dermatite da interface vacuolar, espessamento da membrana basal, edema dérmico, incontinência pigmentar, depósitos de mucina e um infiltrado perivascular composto de linfócitos CD4+. Esses achados são encontrados nas lesões patognomônicas e características da dermatomiosite; entretanto, não são vistas nas “mãos de mecânico”, paniculite, vasculite cutânea, urticária, eritema flagelado e hiperceratose folicular.
A dermatomiosite é distúrbio heterogêneo com vários fenótipos, incluindo miosite, dermatite e doença pulmonar intersticial. Autoanticorpos específicos para miosite identificados recentemente foram associados a características clínicas distintas. Esses autoanticorpos são altamente específicos para doença. Por exemplo, os anticorpos da proteína 5 associada à diferenciação antimelanoma (MDA5) têm alta especificidade para DM clinicamente amiopática, com doença pulmonar rapidamente progressiva. O anticorpo antifator intermediário transcricional 1γ (TIF1), encontrado em pacientes com DM juvenil e adulto, está intimamente relacionado com neoplasias, especialmente em pacientes idosos. Os pacientes com anticorpos anti‐aminoacil‐transferência de RNA sintetase (ARS) compartilham sintomas clínicos característicos, incluindo miosite, doença pulmonar, artrite/artralgia, fenômeno de Raynaud e febre; assim, o termo “síndrome anti‐sintetase” também é usado (tabela 2).10
Autoantígenos e os fenótipos da dermatomiosite
| Autoantígeno | Manifestação clínica |
|---|---|
| MDA5 | DM amiopática; doença intersticial pulmonar |
| TIF1 | DM juvenil; DM associada a neoplasia |
| Mi2 | DM clássica |
| ARS | Síndrome antissintetase; doença pulmonar intersticial crônica |
| NXP2 | DM juvenil e adulta |
| SAE | DM amiopática; miosite grave |
A artrite reumatoide (AR) é doença autoimune caracterizada por inflamação e desenvolvimento de deformidades articulares e pela associação com o fator reumatoide (75% dos pacientes com AR e em 5 a 10% de indivíduos saudáveis) e com o anticorpo antipeptídio citrulinado cíclico de segunda geração (CCP) (especificidade de 90 a 95%).11
As lesões não têm histopatologia típica de AR, mas sugestivas; algumas são específicas da doença e outras apresentam maior associação. São elas: lesões neutrofílicas – pioderma gangrenoso e dermatose neutrofílica reumatoide; granuloma de paliçada – nódulo reumatoide, dermatite granulomatosa e neutrofílica de paliçada, dermatite granulomatosa intersticial; lesões vasculares – vasculite cutânea de pequenos e médios vasos, lesões de Bywater, eritema elevatum diutinum, petéquias e eritema palmar e subungueal; alterações ungueais – onicorrexe, onicomadese, onicólise, baqueteamento digital, lúnula vermelha e pterígio ungueal inverso; outras alterações – amiloidose e urticária crônica espontânea.11
Os nódulos subcutâneos relacionados à AR incluem os nódulos reumatoides clássicos, a nodulose reumatoide acelerada e a nodulose reumatoide.11
Nódulos reumatoides clássicos: lesões nodulares subcutâneas endurecidas únicas ou múltiplas, de tamanho variável, localizadas preferencialmente nas superfícies extensoras dos antebraços e cotovelos. Ocorre em 20 a 30% dos casos; 90% estão associados à presença do fator reumatoide em altos títulos, porém sem relação com gravidade ou progressão da doença.11 Tabagistas têm maior risco de desenvolvê‐los. Também podem ser observados sobre articulações dos dedos, região sacra, tuberosidade isquiática, tendão de Aquiles, região pré‐auricular e couro cabeludo. Quando de grandes dimensões, podem causar compressão nervosa, ulcerar e ser cosmeticamente desagradáveis. Histopatologicamente, exibem grandes nódulos na derme profunda e hipoderme, compostos por fibrina e circundados por granulomas de paliçada.
Nodulose reumatoide acelerada: caracterizada por nódulos subcutâneos prévios (preferencialmente sobre articulações das mãos) que crescem súbita e rapidamente e são independentes da atividade da doença articular.11 Podem ser associadas ao tratamento da AR, preferencialmente o metotrexato.
Nodulose reumatoide: múltiplos nódulos reumatoides subcutâneos sem a presença de AR grave e sem manifestações sistêmicas, mais comum em homens de meia‐idade (30 a 50 anos), autolimitados e benignos. Também há relatos em crianças, sem associação com desenvolvimento de AR, com lesões em áreas atípicas como couro cabeludo, pré‐tibial, joelhos, pés e perimaleolar.
As manifestações vasculares da AR não são específicas. Seu protótipo é a vasculite reumatoide, que consiste numa vasculite leucocitoclástica de pequenos e médios vasos, por depósitos de imunocomplexos, que acomete tanto a pele quanto os vasos mesentéricos, sistema nervoso central e coração, em 2 a 5% dos pacientes com doença de longa data, erosiva e com altos títulos de fator reumatoide. Dependendo do calibre e da localização do vaso acometido, ela pode se manifestar como uma lesão macular ou papular purpúrica, púrpura retiforme, livedo reticular, nódulos subcutâneos e úlceras (geralmente perimaleolares). Infartos digitais secundários ao fenômeno de Raynaud também podem ser observados. A vasculite cutânea de pequenos vasos também pode ocorrer como um fenômeno reacional, em vigência de atividade da doença. O que diferencia a vasculite cutânea da vasculite reumatoide é que a segunda geralmente tem caráter súbito e extenso, com acometimento conjunto de órgãos internos e alta taxa de mortalidade, enquanto a primeira segue curso mais benigno. A neuropatia periférica no contexto da vasculite reumatoide sinaliza pior prognóstico e deve ser rapidamente tratada.
Também são relatadas lesões granulomatosas e neutrofílicas, como a dermatite neutrofílica reumatoide, a dermatite granulomatosa e neutrofílica de paliçada, a dermatite granulomatosa intersticial com artrite, o pioderma gangrenoso e a síndrome de Sweet. Dentro do grupo das lesões granulomatosas, a dermatite granulomatosa neutrofílica é manifestação suspeita de AR, porém rara. Ela se manifesta por pápulas eritematosas simétricas urticariformes, placas, nódulos ou bolhas em mulheres (2:1) com doença erosiva e altos títulos de FR (fator reumatoide), nas superfícies extensoras das articulações (principalmente dorso das mãos e antebraços). A histopatologia revela infiltrado neutrofílico, leucocitoclasia e edema endotelial sem necrose fibrinoide (isto é, sem evidência de vasculite). A espongiose intensa pode causar lesão clínica de vesícula/bolha. Seu curso segue o da atividade da AR.
Existe discussão se as outras lesões granulomatosas estão dentro do espectro das dermatites granulomatosas de paliçada, porém com algumas diferenças clínicas e histopatológicas. Também podem ser chamadas de dermatite granulomatosa intersticial com artrite, granuloma de Churg‐Strauss, pápulas reumatoides, necrobiose reumatoide ulcerada e granuloma necrotizante extravascular de Winkelmann. A dermatite granulomatosa em paliçada se manifesta com pápulas e nódulos normocrômicos dolorosos nas superfícies extensoras dos membros, com umbilicação ou crosta central. Já foram descritas lesões urticariformes e anulares, além de livedo reticular.
A dermatite granulomatosa intersticial com artrite apresenta‐se como pápulas anulares, nódulos e placas eritemato‐violáceas e cordões lineares subcutâneos, simétricos na lateral do tronco e também vistos na face interna das coxas, em mulheres de meia‐idade com FR elevado. Não é exclusivo da AR, e pode ocorrer em outras colagenoses. Pode também estar associada a neoplasia maligna linfoproliferativa subjacente e infecção pelo HIV. A dermatite granulomatosa induzida por medicamentos (hipolipemiantes, anti‐histamínicos anticonvulsivantes e medicamentos anti‐TNF‐α)11 pode ser clinicamente semelhante, porém apresentar dermatite de interface vacuolar. A histopatologia evidencia basicamente infiltrado histiocitário com orla em paliçada, centro com degeneração do colágeno e infiltrado neutrofílico. Na dermatite granulomatosa de paliçada, além dos achados histopatológicos acima, pode haver vasculite leucocitoclástica.
Síndrome de Felty: manifestação extra‐articular rara da AR soropositiva, caracterizada pela tétrade artrite, neutropenia e esplenomegalia. As lesões cutâneas que a acompanham são os nódulos reumatoides (por se tratar de doença erosiva de longa data com elevados títulos de FR) e as úlceras de perna. Está associada a mau prognóstico, com taxa de mortalidade de 25% (geralmente por sepse).
Doença de StillA doença de Still (ou artrite idiopática juvenil, AIJ) é doença reumatológica que cursa com artrite de início na infância ou adolescência. Atualmente, é classificada em sete subtipos.12 O subtipo sistêmico da AIJ é o que cursa com lesões dermatológicas clássicas, que consiste numa erupção macular e papular morbiliforme ou urticariforme evanescente de coloração eritematosa a alaranjada (salmão), que ocorre durante os episódios de febre (usualmente vespertina) e pode preceder ou acompanhar a artrite. A histopatologia não é específica e demonstra infiltrado linfocítico e histiocitário esparso na derme superficial e perivascular.
Existe um tipo de doença de Still que se inicia no adulto, geralmente em mulheres antes dos 30 anos, no qual ocorrem as lesões evanescentes descritas anteriormente, e também lesões atípicas de apresentações clínicas diversas, como pápulas urticariformes, pápulas liquenoides, lesões semelhantes a dermografismo (fig. 4), semelhantes à dermatomiosite, ao prurigo pigmentosa e ao líquen amiloidótico. Todas essas manifestações têm a mesma histopatologia (focos de paraceratose, queratinócitos necróticos únicos ou múltiplos na camada de Malpighi e infiltrado neutrofílico na derme superior e perivascular; ausência de eosinófilos). Essas lesões persistentes “atípicas” têm frequência estimada em 29 a 78% dos pacientes com doença de Still do adulto e, quando presentes, conferem pior prognóstico. São, portanto, marcadores de gravidade de doença.13 Outras manifestações cutâneas associadas com a doença de Still descritas são: alopecia, dor cutânea, lesões acneiformes, prurido, granulomas não caseosos, lesões semelhantes a eczema, urticária, angioedema, vesicopústulas palmoplantares, eritema discrômico migrans, infiltração generalizada em “casca de laranja” (peau d’orange‐like) igual à mucinose cutânea difusa, e o eritema persistente generalizado.14,15
Fenômeno de Raynaud
O fenômeno de Raynaud é definido pela presença de alteração da cor dos dedos que corresponde ao vasoespasmo (palidez e cianose) seguido ou não de vasodilatação (eritema), em razão da exposição súbita a baixas temperaturas em dias de temperatura amena. Pode ser classificado como primário (com sintomas mais brandos) ou secundário à doença de base – por exemplo, a uma colagenose. Nesse caso, a doença é mais agressiva, com maior risco de ulcerações e necrose das extremidades acrais. Geralmente acomete os dedos, porém já foi descrito nas orelhas e nos mamilos.
Em 2014, Maverakis et al. estabeleceram que o diagnóstico é feito com a presença de três dos critérios a seguir: 1) presença da alteração clínica sugestiva de Raynaud (palidez – cianose – eritema); 2) capilaroscopia normal; 3) exame físico negativo para causas secundárias (ulcerações, gangrena, esclerodactilia, calcinose, fibrose cutânea; 4) ausência de história prévia de colagenose; 5) FAN negativo ou em baixos títulos (1:40).
Diante de um paciente com o fenômeno de Raynaud, deve‐se proceder a realização da capilaroscopia, além de completa investigação por meio de anamnese e exame físico minuciosos e painel de autoanticorpos. Alteração na capilaroscopia em paciente com fenômeno de Raynaud é um marcador de risco para a doença ser secundária a uma colagenose.
Causas associadas a fenômeno de Raynaud secundárioDoenças autoimunes: esclerodermia, LE, síndrome de Sjögren, dermatomiosite, doença mista do tecido conjuntivo, colagenoses indiferenciadas, síndrome de sobreposição.
Doenças vasculares: aterosclerose, síndrome do desfiladeiro torácico, vasculite.
Hematológica/oncológica: síndrome POEMS, paraproteinemias, crioglobulinemias, síndromes paraneoplásicas.
Medicamentos: Simpaticomiméticos, bleomicina, interferon, ergotamina, nicotina, policloreto de vinila (PVC).
Endocrinológicas: hipotireoidismo, hipertireoidismo, tireoidite autoimune.16
Síndrome de SjögrenDoença autoimune de etiologia desconhecida, pode ser classificada como primária (SSp) ou secundária (SSs), se associada a outra colagenose. As manifestações cutâneas da SSp são subestimadas, de forma que muitas vezes o dermatologista não faz este diagnóstico. Não existem manifestações cutâneas específicas da SS.17 Por isso é difícil fechar um diagnóstico conclusivo de SSp, já que a doença compartilha características clínicas e imunológicas inespecíficas com as outras colagenoses.18 As manifestações cutâneas são: 1) síndrome sicca: xerostomia, xeroftalmia e xerose; 2) fenômeno de Raynaud; 3) vasculite cutânea: pequenos vasos (IgG/IgM), crioglobulinemia e eritema elevatum diutinum (raro); 4) lesões anulares antiRo (SSA) positivo; 5) amiloidose nodular cutânea localizada; 6) prurido; 7) fotossensibilidade; 8) dermatoses associadas: alopecia (areata), vitiligo, líquen plano, anetodermia, síndrome de Sweet, paniculite granulomatosa, eritema multiforme‐símile, eritema discrômico perstans‐símile e eritema nodoso‐símile.17,18
A síndrome sicca é a manifestação clínica do processo autoimune contra as glândulas salivares, lacrimais e écrinas. Ocorre mais tardiamente na doença, geralmente em indivíduos com idade mais avançada; 50% dos indivíduos com SSp têm xerose.18 Há redução da sudorese com ressecamento cutâneo associado a sintomas como prurido, pinicação e ardência.17 Há alguns relatos de casos de prurido no canal auditivo por redução da produção de cerúmen e dermatite por ressecamento das pálpebras. Sintomas genitais da síndrome sicca incluem dispareunia e prurido vaginal.
O ressecamento ocular e oral ocorre em 85% dos pacientes com SSp.18 A xerostomia causa dor e queimação na mucosa oral, disfagia, queilite angular e maior incidência de cáries.
O fenômeno de Raynaud ocorre em 30% dos casos de SSp e pode ser a primeira manifestação da doença.
A vasculite cutânea ocorre em 10% da SSp, e em 85% dos casos pode ser observada antes do início dos sintomas de síndrome sicca.18 É uma vasculite leucocitoclástica de pequenos vasos e, quando associada a crioglobulinemia, marca mau prognóstico (doença sistêmica mais grave, associação com linfoma B e maior taxa de mortalidade). Clinicamente, pode se manifestar como lesões típicas purpúricas, mas também como lesões urticariformes da urticária vasculite e raramente como eritema elevatum diutinum.18 A púrpura hipergamaglobulinêmica de Waldenström também ocorre no contexto da síndrome de Sjögren e será abordada adiante, na seção das púpuras.
Lesões de fotossensibilidade, pápulas e placas anulares eritematosas e eritema anular policíclico podem ocorrer na SSp (9% dos casos) em indivíduos com positividade para o anticorpo anti‐Ro (SSA), sem associação com LE. Entretanto, a diferenciação (clínica e histopatológica) dessas lesões com as lesões do lúpus subagudo que apresente apenas doença cutânea é muito difícil. As lesões cutâneas estão associadas a menor risco de doença glandular e sistêmica e a melhor prognóstico.18
As lesões podem apresentar infiltrado inflamatório composto por linfócitos e plasmócitos com ou sem redução de glândulas e estruturas ductais écrinas, e infiltrado linfocitário perivascular.17 A imunofluorescência direta demonstra depósitos epidérmicos de IgG intercelular em 2/3 dos casos de SSp e em cerca de 13% de SSs.17
A amiloidose nodular cutânea localizada é uma manifestação rara, e em 25% dos casos está associada à SSp. Localiza‐se geralmente nas pernas e nos braços, e também no tronco e na face. Pode acompanhar amiloidose renal e do trato respiratório inferior.18
EsclerodermiaEsclerodermia é um termo que agrega série de doenças que se manifestam dermatologicamente com espessamento da pele. Ela pode pertencer a síndrome com manifestações vasculares, pulmonares, cardíacas e renais (escleroses sistêmicas) ou ser localizada apenas na pele (esclerodermias localizadas).
A esclerose sistêmica é classificada, segundo o último consenso ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) de 2013, em: esclerose sistêmica cutâneo limitada e esclerose sistêmica cutâneo difusa.19 A seguir, listamos suas características principais:
ES cutâneo limitada: fenômeno de Raynaud; endurecimento da pele das extremidades distais dos membros, face (microstomia) e pescoço; telangiectasias; presença do anticorpo anticentrômero e maior risco de hipertensão pulmonar;
ES cutâneo difusa: início recente de fenômeno de Raynaud; endurecimento da pele da porção proximal dos membros e do tronco; evolução precoce para envolvimento cardíaco; pulmonar (fibrose), renal e gastrintestinal; atrito tendinoso; maior risco de crise renal esclerodérmica; presença do anticorpo anti‐Esclero70 (anti‐topoisomerase), que aumenta o risco de fibrose pulmonar; presença do anticorpo anti‐RNApolimerase III, que confere maior probabilidade de crise renal e câncer.
A histopatologia da pele acometida é característica e demonstra diminuição dos espaços entre os feixes colágenos, aumento de espessura dos feixes colágenos, sem aumento do número de fibroblastos. Também há enclausuramento das glândulas écrinas pela esclerose, que perdem seu coxim adiposo. A alteração se localiza preferencialmente na derme reticular.
As manifestações cutâneas inespecíficas relacionadas à esclerose sistêmica são: capilaroscopia com padrão SD (sclerosis pattern) em 83 a 98% dos casos e de surgimento precoce; telangiectasias; calcinose cutis; discromias: hipercromia semelhante à doença de Addison, máculas hipo e hiperpigmentadas e leucodermia em “sal e pimenta” (que poupa a região perifolicular).20
A esclerodermia localizada (também chamada de morfeia) é a esclerose de qualquer profundidade da pele, e não evolui para a esclerose sistêmica; 20 a 80% dos indivíduos com morfeia podem ter anticorpo FAN positivo, e este não se associa a maior risco de doença sistêmica ou a outra colagenose. Não há alteração dos capilares periungueais na esclerodermia localizada.
Doenças neutrofílicasSíndrome de SweetA síndrome de Sweet se caracteriza clinicamente como lesões eritematosas acompanhadas por febre, artralgias/mialgias e leucocitose, que podem ser recorrentes.
A lesão característica (e histopatologicamente específica da doença) é uma placa eritematosa bem definida de superfície pseudovesicular, vesicular ou pustulosa (fig. 5); menos comumente, podem ser observadas lesões únicas (especialmente em crianças) e lesões nodulares profundas. Esses nódulos podem apresentar um leve eritema sobrejacente e são chamados de Sweet subcutâneo. Raramente os nódulos ulceram e, nesses casos, lembram o aspecto clínico de lesões de pioderma gangrenoso. Sua distribuição predominante é na face, no tronco e nas extremidades proximais; porém, pode ocorrer em áreas de trauma, caracterizando o fenômeno de patergia.21 É mais frequente em mulheres entre 30 a 60 anos.

Em alguns casos, é acompanhada de um quadro gripal prodrômico. Podem ser observados sintomas extracutâneos, como osteomielite estéril, conjuntivite, ceratite ulcerativa e infiltração neutrofílica dos pulmões simulando pneumonia, coração, sistema nervoso, rim e sistema gastrintestinal.
Ela pode ser subdividida nos tipos clássica, parareumática e paraneoplásica. Também pode ser relacionada a gravidez, uso de medicamentos e infecção por HIV. A hipótese de paraneoplasia deve ser aventada na presença de qualquer manifestação atípica – por exemplo, presença de lesões aftoides na mucosa oral, recidiva durante desmame de corticoide, níveis elevados de VHS que não reduzem com a corticoterapia e na associação com doenças reumatológicas.
Os critérios diagnósticos histopatológicos incluem a presença de infiltrado neutrofílico difuso na derme, edema e fragmentação de neutrófilos. Embora o infiltrado possa ser mais pronunciado nas áreas perivasculares, a vasculite está classicamente ausente. As células predominantes do infiltrado dérmico são neutrófilos maduros, embora eosinófilos tenham sido observados em alguns pacientes com síndrome de Sweet clássico ou induzido por medicamentos. No Sweet histiocitoide, as lesões clínicas têm conformação mais anular e, histopatologicamente, observam‐se células imaturas (mieloides) que se assemelham a histiócitos (parecem estar associada com neoplasia hematológica).21
Os principais diagnósticos diferenciais são farmacodermias, eritema polimorfo e hanseníase (sua histopatologia se assemelha à reação hansênica).
Doença de BehçetA doença de Behçet é vasculite multissistêmica caracterizada por úlceras orais e genitais recorrentes, além de manifestações orais, articulares, vasculares, intestinais e do sistema nervoso.22 Ela ocorre em ambos os gêneros e acomete principalmente adultos jovens.
Não existem exames laboratoriais patognomônicos ou achados clínicos específicos. O diagnóstico é feito na presença do critério maior (aftose oral recorrente) associado a dois dos quatro critérios menores (quais sejam: úlcera genital recorrente; lesões oculares – uveíte ou vasculite retiniana; lesões de pele – eritema nodoso, pseudovasculite, lesões papulopustulares ou nódulos acneiformes consistentes com Behçet; e teste de patergia positivo).23
A uveíte posterior (ou seja, vasculite retiniana) é a forma ocular mais clássica observada, embora vários outros achados oculares possam ocorrer. A artrite observada em pacientes com doença de Behçet é não erosiva e inflamatória e afeta tanto grandes quanto pequenas articulações. As manifestações neurológicas têm início tardio nos pacientes e são notavelmente variáveis em sua apresentação. Uma vasculite de vasa vasorum, com tendência a afetar grandes artérias e veias, pode ser uma causa de morte nesses pacientes. Trombose de vasos e aneurismas provavelmente em decorrência de danos endovasculares crônicos são relatados tipicamente como uma manifestação tardia da doença. Os rins são relativamente poupados nos pacientes de Behçet em comparação com outras vasculites.
As lesões mucosas são as lesões ulceradas aftoides. Em geral, são o primeiro sintoma da doença (podem preceder os outros sintomas por anos) e também ocorrem durante atividade da doença. Úlceras genitais no pênis, na bolsa escrotal e na vulva são dolorosas, têm fundo de fibrina e demoram para cicatrizar (fig. 6).
.")
Na pele, as lesões são vesicopústulas estéreis ou papulopurpúricas (não foliculocêntricas) acrais e faciais. Lesões de paniculite (eritema nodoso‐símile e histologicamente semelhantes à vasculite nodular) nas pernas e nádegas ocorrem principalmente nas mulheres. Lesões de eritema nodoso propriamente dito e lesões de tromboflebite também ocorrem no contexto da doença de Behçet. O fenômeno de patergia é descrito, assim como no pioderma gangrenoso.
A histopatologia é inespecífica. A lesão acneiforme pode ser uma vasculopatia com trombose e vasculite neutrofílica, e infiltrado histiocitário de permeio. As lesões mais antigas têm predomínio de linfócitos. As lesões nodulares subcutâneas são uma paniculite lobular com ou sem comprometimento septal, necrose de adipócitos, além dos achados vasculares descritos.
Síndrome artrite‐dermatite associada ao trato gastrintestinalA síndrome artrite‐dermatite associada ao trato gastrintestinal é caracterizada por surtos recorrentes de lesões pustulosas (foliculares e não foliculares) e abscessos, sinovite, febre e sintomas gripais que se desenvolvem após procedimentos cirúrgicos do trato gastrintestinal (frequentemente cirurgia bariátrica) ou em pacientes com doença inflamatória intestinal.24
As lesões são recorrentes (a cada 4 a 6 semanas). Podem ser observadas máculas, pápulas e vesico‐pústulas eritematosas e purpúricas, nas extremidades proximais e no tronco. Também nódulos subcutâneos recorrentes dolorosos e eritematosos (é uma paniculite neutrofílica lobular) no tronco e nas extremidades, que curam com cicatrizes atróficas deprimidas. O eritema nodoso verdadeiro também é uma manifestação da doença.
Na histopatologia, as lesões iniciais não são específicas e os achados são muito semelhantes às lesões da doença de Behçet. Há edema papilar e vesículas subepidérmicas nas lesões mais recentes; enquanto lesões mais tardias exibem denso infiltrado dérmico neutrofílico.
Pioderma gangrenosoPioderma gangrenoso é dermatose neutrofílica rara que se caracteriza por úlceras cutâneas rapidamente progressivas. Essas úlceras se expandem perifericamente, são dolorosas e apresentam bordas bem definidas de coloração eritematosa a violácea (fig. 7). Pode ser observado o fenômeno de patergia e, quando regride, a cicatriz atrófica adquire um padrão cribriforme. Por ter histopatologia inespecífica, consiste em diagnóstico de exclusão. A tabela 3 descreve as condições sistêmicas associadas ao pioderma gangrenoso.25
. À esquerda: úlcera profunda e fundo contendo fibrina na periferia, sem borda arroxeada (lesão mais antiga, em área de cicatriz de cesariana).")
Pioderma gangrenoso. À direita: lesão ulcerada com margens irregulares, inflamatórias e elevadas, de cor vermelho‐escura ou purpúrica e base necrótica (lesão em atividade do pioderma gangrenoso). À esquerda: úlcera profunda e fundo contendo fibrina na periferia, sem borda arroxeada (lesão mais antiga, em área de cicatriz de cesariana).
Doenças associadas ao pioderma gangrenoso
| Hematológicas | Mielofibrose |
| Leucemia mielocítica | |
| Metaplasia mieloide agnogênica | |
| Leucemia de células pilosas | |
| Gamopatia monoclonal por IgA | |
| Policitemia vera | |
| Mieloma múltiplo | |
| Reumatológicas | Artrite soronegativa com doença inflamatória intestinal |
| Soronegativa sem doença inflamatória intestinal | |
| Artrite reumatoide | |
| Osteoartrite | |
| Espondiloartropatias | |
| Lúpus eritematoso sistêmico | |
| Arterite de Takayasu | |
| Trato gastrointestinal | Retocolite ulcerativa |
| Doença de Crohn | |
| Hepatite crônica em atividade | |
| Enterite regional | |
| Cirrose biliar primária | |
| Endocrinológicas | Diabetes mellitus |
| Tireoidopatias | |
| Outras | Síndromes autoinflamatórias com: |
| Hidradenite supurativa | |
| Dermatoses neutrofílicas | |
| Acne conglobata | |
| Hemoglobinúria paroxística noturna | |
| Sarcoidose | |
| Neoplasias | |
| Pneumopatias |
Quaisquer doenças que se manifestam com úlceras entram no diagnóstico diferencial de pioderma gangrenoso, como a leishmaniose e a esporotricose. Nos casos clinicamente semelhantes ao pioderma gangrenoso, mas que se comportam de maneira distinta, devemos aventar a possibilidade de se tratar de granulomatose com poliangiíte. Além disso, num pioderma gangrenoso refratário à terapêutica tradicional, deve‐se pensar em doença autoinflamatória.
O pioderma gangrenoso apresenta algumas variantes:
- ‐
Pioestomatite vegetante: erosões crônicas, pustulosas e eventualmente vegetantes nas membranas mucosas;
- ‐
Pioderma gangrenoso atípico ou bolhoso: ulcerações mais superficiais nas extremidades superiores e face. Descrito em doenças hematológicas como síndrome mielodisplásica ou leucemia mieloide aguda;
- ‐
Pioderma gangrenoso periostomal.
É vasculite leucocitoclástica cutânea rara que se apresenta inicialmente como pápulas eritemato‐violáceas que coalescem para formar placas eritemato‐amareladas lembrando lesões urticarifomes. As lesões tendem a surgir em áreas de trauma e a respeitar uma distribuição simétrica nas superfícies extensoras das articulações das extremidades e nos glúteos. O quadro pode estar associado a artralgias e artrite.
As malignidades hematológicas são consideradas os fatores mais comumente associados, das quais a mais frequente é a gamopatia monoclonal por IgA.26
Também é relatada sua associação com doenças neoplásicas, autoimunes e infecciosas, destacando‐se a infecção pelo HIV, em que a apresentação clínica pode ser alterada com presença de lesões nodulares e envolvimento palmoplantar.27
PúrpurasAs púrpuras podem se associar tanto a doenças graves e potencialmente fatais quanto a condições crônicas comuns e benignas. Em ambos os casos, elas se caracterizam como manifestações não específicas de uma miríade de enfermidades. De qualquer modo, na maioria das vezes uma boa anamnese e exame clínico associados a alguns exames laboratoriais são suficientes para sua avaliação.
A morfologia da púrpura auxilia tanto em sua distinção quanto no mecanismo fisiopatogênico envolvido. As púrpuras podem ser resultantes de um processo hemorrágico simples, uma hemorragia secundária a inflamação dos vasos e, por fim, uma hemorragia oclusiva com secundária inflamação mínima.
Hemorragia simples petequialTrombocitopenia ou disfunção plaquetária: podem ser observadas equimoses, mas a morfologia predominante é a petéquia. As petéquias podem surgir com contagem plaquetária <50.000 mm3; entretanto, são tipicamente observadas com contagem plaquetária ≤ 10.000 mm3. A trombocitopenia grave resulta na deterioração da junção endotelial dos vasos levando a um aumento da permeabilidade vascular com extravasamento de hemácias.
Já os distúrbios da função plaquetária levam mais frequentemente a equimoses secundárias a mínimos traumas do que ao surgimento de petéquias.28
Picos de pressão intravascularPicos de pressão intravascular fortes ou repetitivos podem resultar em hemorragia petequial, como o observado acima das clavículas após o trabalho de parto, acessos de tosse ou vômitos e choro excessivo em crianças. O uso de garrotes pode levar a uma distribuição local de petéquias (acima do local comprimido).
Síndromes microvasculares minimamente inflamatóriasAs púrpuras pigmentares crônicas e a púrpura hipergamaglobulinêmica de Waldenström acometem vasos menos calibrosos da derme. Na primeira, observam‐se área de hemorragia recorrente, frequentemente petequial com eritema adjacente e hiperpigmentação acastanhada como resultado do depósito de hemossiderina, o que resulta no aspecto residual de coloração ocre. Dependendo do aspecto clínico ou do processo inflamatório subjacente, pode ser denominada púrpura de Schamberg (apresenta depósitos de hemossiderina puntiformes, com aspecto de “pimenta caiena”), púrpura telangiectásica de Doucas e Kapetanakis (máculas ou pápulas descamativas purpúricas), púrpura anular telangiectoide de Majocchi (lesões purpúricas de formato anular), líquen aureus (placa única purpúrica a castanhada ou, se mais antiga, ocre), e, mais raramente, as variantes púrpura pigmentar linear e púrpura pigmentar granulomatosa.
A hipergamaglobulinêmica de Waldenström é caracterizada por hemorragia macular em áreas dependentes de gravidade e áreas cobertas por vestimentas apertadas, e pode ser acompanhada de queimação local. Ela pode ser idiopática ou se associar a patologias que cursam com gamopatia policlonal, como síndrome de Sjögren, sarcoidose e outras doenças.29
Problemas na cascata de coagulaçãoCausas vascularesCausas inflamatórias: nesses casos, é observada inflamação direcionada aos vasos. A inflamação perivascular não é considerada vasculite e não resulta em púrpura palpável. Se as lesões forem secundárias a depósitos de imunocomplexos, elas podem ser comumente observadas em áreas dependentes de gravidade. Uma causa importante de púrpura palpável é a vasculite leucocitoclástica, que pode ser observada em diversas patologias (descritas adiante).
Causas não inflamatórias: púrpuras retiformes (arboriforme, estrelada) dolorosas, de apresentação aguda, são típicas da síndrome do anticorpo antifosfolipídio e de outras vasculopatias trombóticas. Costumam se manifestar nas extremidades, hélices auriculares, bochechas, fronte e tronco, ou podem ser disseminadas. Ocasionalmente, algumas lesões podem ter eritema na periferia e bolhas necróticas. No exame histopatológico, observa‐se trombose difusa não inflamatória dos vasos da derme, sem evidência de vasculite.30
Causas extravascularesGrandes traumas podem resultar em hemorragias cutâneas. Nesses casos, edema, dor e escoriações sugerem o diagnóstico. Já as púrpuras relacionadas a mínimos traumas costumam ocorrer como resultado de um tecido conjuntivo que oferece pouco suporte aos pequenos vasos da pele, como observado na púrpura actínica, por excesso de corticosteroides (locais ou sistêmicos), na síndrome de Ehlers‐Danlos, no pseudoxantoma elástico e na amiloidose de cadeias leves. Nesta última, as púrpuras são facilmente induzidas pelo pinçamento da pele.
A presença de hemorragias perifoliculares sugere o diagnóstico de escorbuto.
VasculitesA vasculite é consequência da inflamação dos vasos sanguíneos.31 Pode representar uma doença limitada à pele, uma manifestação de doença sistêmica ou medicamento, ou uma enfermidade primária da pele com repercussão sistêmica. Podem ser causadas por infecções, medicamentos, neoplasias, doenças autoimunes e do tecido conjuntivo. Nesses casos, constituem uma alteração inespecífica ao agente causal. Entretanto, aproximadamente metade dos casos é vasculite leucocitoclástica idiopática.
As repercussões clínicas dependem da localização e do tamanho do vaso acometido.32
O Consenso da Conferência Internacional de Chapel Hill, em 2012, definiu a nomenclatura das vasculites sistêmicas.32 São classificadas em:
Vasculites primáriasDe pequenos vasos: relacionadas ao ANCA (poliangiíte microscópica, granulomatose com poliangiíte, granulomatose eosinofílica com poliangiíte) e associadas aos depósitos de imunocomplexos (vasculite por IgA, vasculite crioglobulinêmica, vasculite‐urticária hipocomplementêmica, antimembrana basal glomerular.
Vasculites de médios vasos: poliarterite nodosa e doença de Kawasaki.
Vasculite de grandes vasos: arterite de Takayasu.
Vasculites de vasos de tamanhos variáveis: doença de Behçet e síndrome de Cogan.
Vasculite de órgão único: vasculite leucocitoclástica limitada à pele, arterite cutânea, aortite isolada, vasculite primária do sistema nervoso central.
As vasculites cutâneas podem se manifestar com alteração de coloração na pele como eritema, petéquias e livedo reticular e até dano celular em todas as camadas, como ocorre na gangrena digital.
Quando são acometidos os pequenos vasos, geralmente localizados na derme papilar, provocam um exantema maculopapular que evolui para púrpura palpável. Contudo, essas lesões não desaparecem com a pressão. Também estão descritas máculas, petéquias, vesículas, bolhas e ulcerações.31
O dano aos vasos médios, que se localizam na derme profunda, causa lesões maiores e mais profundas. Estão descritos nódulos, úlceras, livedo e lesões necróticas. Na pele, não temos grandes vasos.31
A apresentação clínica mais comum das vasculites cutâneas é a púrpura palpável, e a região distal dos membros inferiores é a área mais predominante. As principais manifestações extracutâneas presentes são: artralgias e artrite, alteração renal e envolvimento gastrintestinal.
Arterite de TakayasuA arterite de Takayasu é vasculite de grandes vasos; contudo, pode acometer vasos menos calibrosos. A inflamação evolui para um estágio de oclusão que pode levar a estenose ou formação de aneurismas. É mais comum nas mulheres e geralmente inicia na segunda e terceira décadas de vida. Na pele, pode causar púrpura, livedo reticular, lesões subcutâneas semelhantes ao eritema nodoso, eritema indurado, nódulos inflamatórios, nódulos necróticos e/ou ulcerados, gangrena digital, úlceras semelhantes ao pioderma grangrenoso, fenômeno de Raynaud, urticária, angioedema e eritema multiforme.32,33 Os nódulos não costumam ter relação com o local de comprometimento vascular e podem surgir em qualquer fase da doença. Foram descritos casos de associação com síndrome de Sweet em crianças.33
Arterite de células gigantesAs manifestações cutâneas da arterite de células gigantes são raras. Podem ser lesões isquêmicas consequentes a obstrução da artéria ou lesões geradas por outros mecanismos. Foram descritos induração, eritema e bolha no couro cabeludo e têmporas, glossite e necrose da língua, necrose do couro cabeludo, púrpura, gangrena distal dos membros, equimose periorbitária, edema na face e no pescoço, nódulos, granuloma anular.33
Poliarterite nodosaA poliarterite nodosa (PAN) sistêmica é vasculite necrotizante de artérias de médio ou pequeno calibre sem glomerulonefrite ou vasculite de arteríolas, capilares ou vênulas não associada ao ANCA. Podemos encontrar púrpura palpável, livedo reticular, nódulos, lesões urticariformes, eritema transitório, flebite superficial, necrose distal e hemorragia por estilhaço.33
A PAN cutânea é vasculite necrotizante de arteríolas da derme profunda e subcutâneo. Pode estar relacionada a infecções (hepatite B, hepatite C, M. tuberculosis, HIV, HTLV, parvovirus B19, citomegalovirose; e nas crianças, estreptococose), doenças inflamatórias, artrite reumatoide, fármacos (penicilina, minociclina). Os principais achados são nódulos subcutâneos, livedo reticular, púrpura e úlceras.33 Sintomas como febre, perda de peso, artrite e neuropatia podem surgir em qualquer apresentação.
Doença de KawasakiA doença de Kawasaki é vasculite aguda que ocorre principalmente em crianças. Geralmente o quadro começa com febre e exantema polimórfico de disseminação centrífuga, além de enantema orofaríngeo e língua em morango. Após alguns dias, surge descamação perineal e nas pontas dos dedos.33 A doença pode evoluir com vasculite e formação de aneurismas nas artérias coronárias. Dessa maneira, a suspeição do diagnóstico é fundamental para o início precoce do tratamento e a minimização de sequelas.
Vasculites associadas ao ANCAAs vasculites associadas ao ANCA causam mais comumente a púrpura palpável; contudo, podem eventualmente apresentar livedo racemoso, eritema nodoso, nódulos subcutâneos e úlceras dolorosas. A vasculite leucocitoclástica com predomínio de neutrófilos é o achado histopatológico mais comum na poliangiíte microscópica e na granulomatose com poliangeíte (GPA) – conhecida como granulomatose de Wegener. Já na granulomatose eosinofílica com poliangiíte (chamada previamente de síndrome de Churg Strauss), predominam granulomas necrotizantes extravasculares com infiltrado misto de neutrófilos e eosinófilos.32,33
Vasculites associadas a imunocomplexosA vasculite por IgA, mais conhecida na criança como púrpura de Henoch Schönlein, é a vasculite mais comum da infância e geralmente se apresenta como pápulas eritematosas que surgem de forma ascendente a partir da região distal dos membros inferiores e evoluem para púrpura palpável.
A vasculite crioglobulinêmica é causada pelo depósito de crioglobulinas nos pequenos vasos. De acordo com as crioglobulinas depositadas, podem ser divididas em tipo I (monoclonal), tipo II (mono e policlonal) ou tipo III (policlonal).32 O tipo I está relacionado a desordens linfoproliferativas dos linfócitos B, como o mieloma múltiplo, e se manifesta como uma vasculopatia trombótica (fig. 8). Os tipos II e III estão associados a infecções (principalmente a hepatite C), autoimunidade (artrite reumatoide, síndrome de Sjögren e LES) ou doenças neoplásicas, mas também podem ser idiopáticas.34 A tipo III idiopática é chamado de crioglobulinemia mista essencial. A forma clínica mais comum de crioglobulinemia é o surgimento intermitente de petéquias palpáveis nos membros inferiores. Acrocianose, necroses e úlceras dolorosas também podem aparecer,32 acompanhadas ou não de artralgia, artrite, neuropatia e nefropatia.

A vasculite urticariforme hipocomplementêmica apresenta‐se com placas urticariformes que duram mais que 24 horas, geralmente doem mais do que coçam e deixam pigmentação local. É mais comum nas mulheres e pode vir acompanhada por artralgia e alteração ocular. Para o diagnóstico, devem estar presentes as lesões urticariformes recorrentes e complemento sérico reduzido, bem como duas das seguintes características: vasculite cutânea (leucocitoclástica), artrite ou artralgias, inflamação ocular, glomerulonefrite, dor abdominal e ou anticorpo anti‐C1q. Devem ser investigadas hepatite B, neoplasias, LES e síndrome de Sjögren.
Vasculite de vasos variáveisA doença de Behçet é doença associada a uma vasculite obstrutiva imunomediada que foi abordada no item de doenças neutrofílicas.
A síndrome de Cogan é doença imune de acometimento ocular e auditivo que pode estar associada a vasculite sistêmica.
Vasculites secundáriasAssociada a doenças sistêmicas (LES, vasculite da artrite reumatoide, vasculite da sarcoidose). Vasculites associadas a etiologia provável: neoplasia, infecciosa, medicamentosa (associadas ao ANCA ou por depósitos de imunocomplexos).
A tabela 4 descreve os tipos de vasculites que ocorrem no contexto de algumas doenças sistêmicas.31,32,35–42
Vasculites secundárias
| Lúpus eritematoso | Vasculite leucocitoclástica cutânea: máculas e pápulas eritematosas, púrpura palpável, lesões isquêmicas ou ulceradas, lesões urticariformes e nódulos31,32 |
| Artrite reumatoide | Vasculite leucocitoclástica cutânea: púrpura palpável, úlceras, lesões isquêmicas digitais, eritema maculopapular, bolhas hemorrágicas, eritema elevado diutino, livedo reticular, nódulos subcutâneos e vasculite livedoide31,32 |
| Sarcoidose | Vasculite leucocitoclásica com ou sem granulomas, livedo reticular, úlceras nos membros inferiores e lesões pupúricas ou anulares35,36 |
| Síndrome de Sjögren | Vasculite leucocitoclásica, urticária‐vasculite e vasculite crioglobulinêmica31,37 |
| Eritema elevatum diutinum | Vasculite crônica |
| Medicamentos (resolução com a suspensão) | anti‐TNFα, levamisole, propiltiouracil, hidralazina, rituximabe, montelucaste, estatinas39 |
| Neoplasia | Síndromes paraneoplásicas (doenças linfo e mieloproliferativas) |
| Outras | Dermatomiosite |
| Esclerose sistêmica | |
| Doenças inflamatórias intestinais | |
| COVID‐19 (leucocitoclástica de pequenos vasos cutânea, vasculite urticarial e vasculite linfocitica perniose‐símile)40 |
Tem causa e patogênese desconhecida. Acredita‐se que algum agente ambiental (vírus, bactéria, autoantígenos, sílica, pólen) atue no hospedeiro geneticamente suscetível, ativando a resposta imune, com resultante formação de granuloma. Está presente na pele em 25 a 30% dos casos e, nestes, em 50% atinge algum outro órgão em conjunto.43
As lesões específicas da sarcoidose são aquelas que, ao serem biopsiadas, revelam granulomas epitelioides não caseosos e se caracterizam por pápulas pequenas, eritemato‐violáceas (apresentação mais comum, com localização na cabeça e pescoço, áreas periorbital, perinasal e perioral) semelhantes às de LCC discoide, placas, nódulos, nódulos subcutâneos (Darier‐Roussy: assintomático, no tronco e nas extremidades, tendo ou não epiderme violácea), lesões sobre cicatrizes, pápulas associadas a tatuagem e lúpus pérnio (lesões infiltradas no nariz e mento). Quando o acometimento é extenso, há maior taxa de doença respiratória e cistos ósseos. Pode haver também lesão da mucosa nasal e osso subjacente. É a forma mais refratária ao tratamento. Outras lesões específicas, porém mais raras, são ictiose adquirida, eritrodermia, lesões psoriasiformes, úlceras, lesões verrucosas, alopecia, lesões fotodistribuídas e sarcoidose angiolupoide (lesões de lúpus pérnio com telangiectasias; maior chance de ulceração e associação com doença ocular). A sarcoidose é conhecida como a grande imitadora. A lesão inespecífica é representada pelo eritema nodoso, com acometimento simétrico, na região pretibial, com bom prognóstico. Pode ser acompanhado por uveíte, febre, artrite e linfadenomegalia hilar bilateral, caracterizando a síndrome de Löfgren (forma aguda que se resolve espontaneamente em até 90% dos casos e que pela especificidade não necessita de biópsia para fechar o diagnóstico). Outras síndromes associadas são: Heerfordt‐Waldenström (febre, aumento de parótidas, uveíte anterior e paralisia do nervo facial), Mikulicz (acometimento das glândulas lacrimais, sublingual, submandibular e parótidas) e sarcoidose‐linfoma (desenvolvimento do tipo Hodgkin ou não Hodgkin em portador de sarcoidose crônica; é de suma importância diferenciar de lesões similares à sarcoidose desencadeadas por anticorpos monoclonais usados contra neoplasias).44
Todos os casos de sarcoidose cutânea devem ser investigados para doença sistêmica. O diagnóstico é feito com a junção da clínica, achados radiológicos, laboratório, diascopia (aspecto de geleia de maçã) e a biópsia (histopatologia com granuloma não caseoso). A lesão cutânea é a de mais fácil acesso para a histopatologia. Entretanto, também é indicada a biópsia de linfonodos palpáveis e glândula salivar para avaliar se há comprometimento sistêmico.
A mortalidade gira em torno de 3 a 6%, por envolvimento cardíaco, pulmonar e neurológico.
Necrobiose lipoídicaDoença granulomatosa inflamatória crônica, associada com degeneração do tecido conectivo, comumente vista em associação com diabetes, hipertensão, doenças da tireoide e obesidade. É três vezes mais frequente em mulheres.44
A clínica se apresenta como placas irregulares, ovoides, com a periferia violácea, endurecida e com centro atrófico, amarelo e brilhante. Telangiectasias na superfície são comuns. Pode ter hipo‐hidrose, alopecia e anestesia associadas. É mais frequente na região pré‐tibial; ulceração é comum, mesmo com trauma mínimo.
O diagnóstico é feito com a clínica e a biópsia da borda da lesão, incluindo tecido subcutâneo, que detecta granuloma intersticial em paliçada, composto por histiócitos e células gigantes multinucleadas na derme e subcutâneo, associado a degeneração do colágeno.
Granuloma anularDoença granulomatosa cutânea não infecciosa, associada com diversas doenças, diabetes, doença tireoideana, dislipidemia, infecções (HIV e borreliose) e malignidades, resultando na necessidade de investigação adicional. Parece ser reação mediada por célula a um antígeno desconhecido, com possíveis disparadores, incluindo trauma, medicamento (incluindo imunoterapia contra o câncer), vacinação e luz ultravioleta.44 A clínica pode ser localizada (75% dos casos, com pápulas cor da pele, eritemato‐violáceas ou acastanhadas, não escamativas que coalescem formando placas anulares), generalizada (atinge tronco e extremidades, com maior refratariedade, início mais tardio, e pode ter sensação de queimação) e com forma subcutânea (mais nas pernas em crianças, com diagnóstico diferencial com nódulo reumatoide).45 As formas mais atípicas são: granuloma anular perfurante (ulcerado, umbilicado ou com crosta/escama central), palmoplantar, pustuloso, visceral.
Em geral, é assintomático; o tratamento é feito, na maioria das vezes, por razões cosméticas.46 A investigação deve ser feita com exames laboratoriais, com hemograma, hormônios tireoidianos, hemoglobina glicosilada, lipidograma e sorologias para hepatite B, C e HIV. O rastreio de neoplasia deve ser feito de acordo com a idade.
Suporte financeiroNenhum.
Contribuição dos autoresAna Luisa Sampaio: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Aline Lopes Bressan: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura.
Barbara Nader Vasconcelos: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura.
Alexandre Carlos Gripp: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; participação efetiva na orientação da pesquisa.
Conflito de interessesNenhum.
Agradecemos imensamente a contribuição das Dras Danielle Mello, Gabriela Higino, Juliana Martins Leal e Paula Figueiredo e Marsillac pela ajuda com a confecção deste artigo.
Como citar este artigo: Sampaio AL, Bressan AL, Vasconcelos BN, Gripp AC. Skin manifestations associated with systemic diseases – Part I. An Bras Dermatol. 2021;96:655–71.
Trabalho realizado no Hospital Universitário Pedro Ernesto, Rio de Janeiro, RJ, Brasil.
