









Dermatomiosite juvenil (DMJ) é miopatia rara. A lipodistrofia é complicação subnotificada da DMJ crônica.
ObjetivoAvaliar a evolução a longo prazo da DMJ associada à lipodistrofia.
MétodosEstudo retrospectivo de pacientes com DMJ que desenvolveram lipodistrofia, diagnosticados e tratados entre 1° de janeiro de 1990 e 31 de dezembro de 2023, em clínica de dermatologia universitária. O seguimento médio foi de 150,8 meses (variação de 29 a 291).
ResultadosA lipodistrofia foi diagnosticada em cinco crianças (quatro meninas e um menino, com idades entre 3 e 14 anos) de 22 pacientes com DMJ (17 meninas e cinco meninos). Quatro pacientes apresentavam lipodistrofia parcial e um apresentava lipodistrofia focal que ocorreu 34,2 meses (período médio) após os sintomas iniciais da DMJ. Todas as cinco crianças apresentaram positividade para anticorpos antinucleares, mas nenhuma tinha anticorpos específicos/associados à dermatomiosite. Malignidade ou envolvimento visceral não foram encontrados. Na apresentação da DMJ, todos os cinco pacientes apresentavam vitamina D sérica baixa, e o IMC variou de 14,3 a 20,7. Os níveis de triglicérides e glicemia de jejum eram normais em todos os pacientes, enquanto o colesterol estava elevado em um paciente. Apesar da terapia imunossupressora (TIS) padrão, dois de quatro pacientes com lipodistrofia inicialmente parcial progrediram para lipodistrofia generalizada, e quatro de cinco pacientes desenvolveram calcinose 46 meses (período médio) após o diagnóstico de DMJ. Na última consulta, três de cinco pacientes tinham curso crônico de DMJ, necessitando de TIS, enquanto dois de cinco pacientes estavam em remissão, sem TIS, apenas com lipodistrofia residual.
Limitações do estudoEstudo retrospectivo.
ConclusõesDurante o período de 34 anos, cinco (23%) de 22 pacientes com DMJ desenvolveram lipodistrofia. Não foram encontradas complicações metabólicas. Todos os pacientes tiveram atraso significante no diagnóstico de DMJ. O diagnóstico precoce e TIS são necessários para reduzir o risco de lipodistrofia, complicação grave e crônica da DMJ.
Dermatomiosite (DM) é doença rara e crônica do tecido conjuntivo, que afeta principalmente a pele e os músculos. A DM é de etiologia desconhecida, apresentando‐se como doença do adulto ou na forma juvenil (início antes dos 18 anos). A dermatomiosite juvenil (DMJ) é a miopatia pediátrica mais comum, com incidência estimada de aproximadamente 3,1 crianças por milhão.1 Fraqueza muscular proximal e lesões cutâneas características são manifestações clínicas essenciais, juntamente com sinais de miopatia inflamatória extensora proximal. Antes da era dos glicocorticoides, a DMJ levava à morte 1/3 dos pacientes, mas com o avanço da terapia e do diagnóstico, a mortalidade diminuiu para 2% a 3,1% atualmente relatados.2–4 Embora muitos alcancem a remissão, complicações a longo prazo e danos específicos ainda podem se desenvolver, como lipodistrofia, calcinose, fraqueza residual, contraturas articulares, atrofia muscular, hirsutismo, falha de crescimento e disfagia.4,5
A lipodistrofia é complicação menos frequente, mas significante, da DMJ e pode ser localizada ou generalizada. A lipodistrofia pode ser adquirida ou congênita e pode estar associada a anormalidades metabólicas, como hiperlipidemia, tolerância à glicose prejudicada, diabetes mellitus resistente à insulina, acantose nigricans, hepatomegalia.6,7 A lipodistrofia adquirida (LDA) tem sido associada a doenças autoimunes7 e a LDA infantil tem sido associada à DMJ,7 lúpus eritematoso sistêmico (LES),8 paniculite,9 síndrome de Sjögren e artrite reumatoide juvenil.10,11 Curiosamente, a lipodistrofia está associada quase exclusivamente à DMJ, embora tenham sido publicados relatos de casos de lipodistrofia associada à dermatomiosite de início na idade adulta.12–16 A prevalência de lipodistrofia em pacientes com DMJ varia de 7,93% a 40%.4,5,7,12,17–21 A lipodistrofia na DMJ pode ser categorizada em três fenótipos, dependendo da distribuição da perda de gordura subcutânea: generalizada (perda na face, tronco, abdome e todas as extremidades), parcial (extremidades superiores e/ou inferiores, com preservação relativa do abdome e tronco) ou focal (áreas localizadas, resultando em depressão da superfície da pele ou covinhas).6,22,23
Este relato apresenta quatro meninas e um menino com DMJ que desenvolveram diferentes fenótipos de lipodistrofia. São descritas as manifestações clínicas da DMJ e lipodistrofia associada, análises laboratoriais, complicações e opções de tratamento durante o seguimento a longo prazo.
Materiais e métodosA partir do banco de dados disponível foi conduzida revisão retrospectiva e análise de dados clínicos de pacientes com DMJ confirmada associada à lipodistrofia no Departamento de Dermatologia Pediátrica do University Clinical Center da Sérvia entre 1° de janeiro de 1990 e 31 de dezembro de 2023. A busca nos bancos de dados PudMed e Medline foi realizada usando as palavras‐chave “lipodystrophy” OR “lipoatrophy” AND “juvenile dermatomyositis”. A busca reversa de citações foi realizada e casos adicionais foram identificados.
ResultadosVinte e dois pacientes com DMJ (17 meninas, cinco meninos – proporção de 3,4:1 sexo feminino:masculino) foram diagnosticados e/ou encaminhados para o departamento. Erupção cutânea foi vista em todos os pacientes do presente estudo com DMJ (22/22) como a queixa mais frequente na apresentação inicial, seguida por artralgia, mialgia e fraqueza isoladamente, que foram observadas em 54,5% dos pacientes (12/22), respectivamente, enquanto 36,4% tiveram uma infecção anterior (8/22). Durante período de 34 anos, 477 pacientes adultos com DM (351 do sexo feminino, 126 do sexo masculino – proporção de 2,79:1,0 sexo feminino:masculino) foram diagnosticados. A DMJ representou aproximadamente 4,2% de todos os casos de DM e foi 26,5 vezes menos frequente na presente experiência.
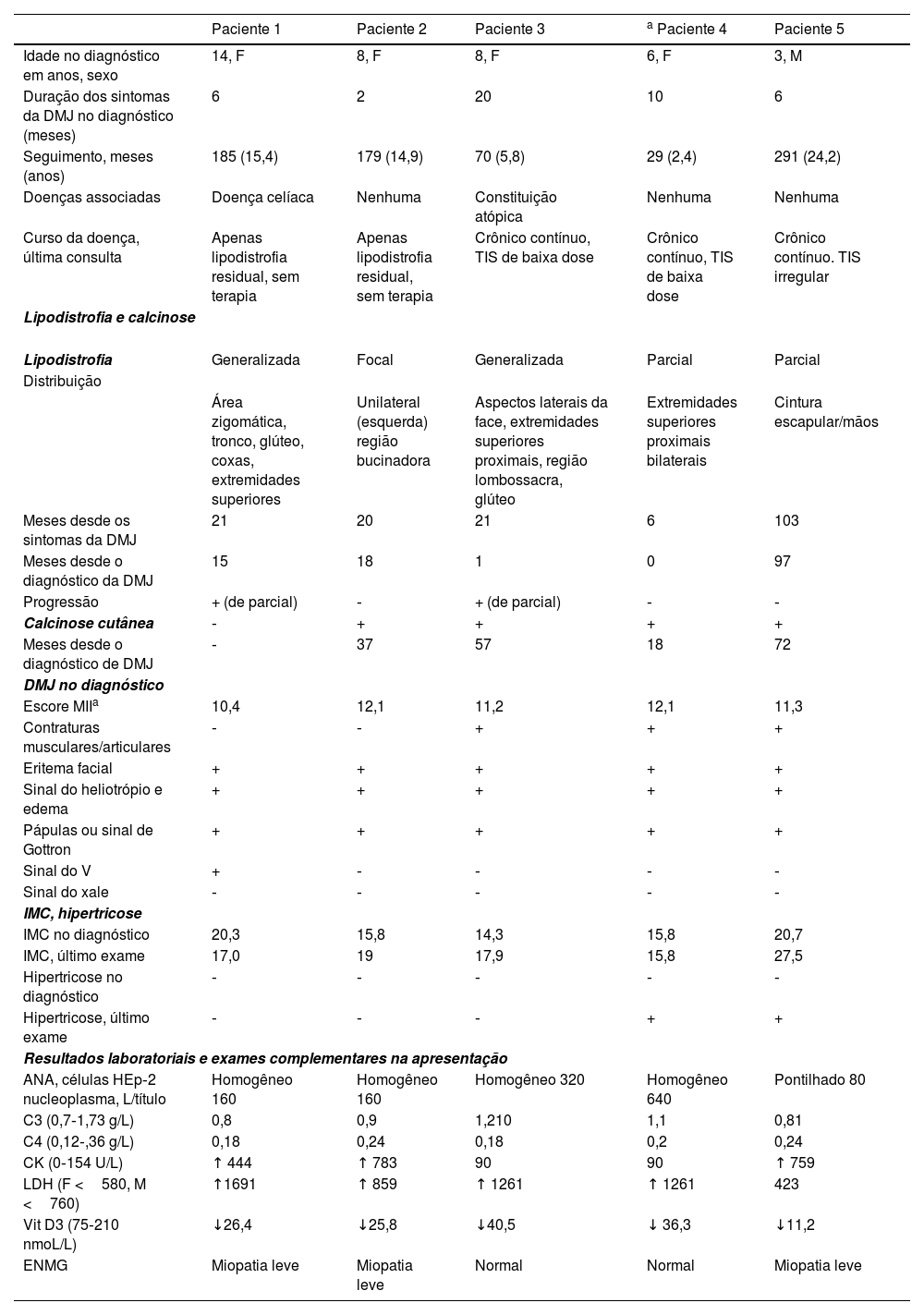
Lipodistrofia foi encontrada em 22,7% dos pacientes com DMJ (5/22; quatro meninas e um menino, com idades entre 3 e 14 anos). O tempo entre os sintomas e o diagnóstico de DMJ foi de 8,8 meses (variação de 2 a 20) e a lipodistrofia ocorreu 34,2 meses (período médio) após os sintomas iniciais de DMJ. O período médio de seguimento foi de 150,8 meses (variação de 29 a 291). De acordo com os critérios de classificação da European League against Rheumatism e do American College of Rheumatology (EULAR/ACR) para miopatias inflamatórias idiopáticas adultas e juvenis e seus principais subgrupos, os pacientes tiveram escore de miopatias inflamatórias idiopáticas (MII) de 10,4, 12,1, 11,2, 12,1 e 11,3, respectivamente, e foram categorizados como definitivos.24 Todos os pacientes foram classificados como crônicos contínuos. Não foram detectados envolvimento de órgãos internos específicos e malignidade durante o seguimento em longo prazo. Todos os cinco pacientes tinham níveis de triglicérides, lipoproteína de alta densidade (HDL), lipoproteína de baixa densidade (LDL) e glicemia de jejum normais na apresentação da lipodistrofia. Apenas um de cinco dos pacientes tinha níveis elevados de colesterol, 6,38 (v.n.<5,20 mmoL/L). Nenhum paciente tinha acantose nigricans na apresentação e durante o seguimento. Todas as crianças tinham anticorpos antinucleares (ANA; tabela 1), mas nenhuma tinha anticorpos específicos para miosite (MSA, do inglês myositis‐specific autoantibodies; anti‐Jo, anti PL‐7, anti PL‐12, anti Mi‐2 e anti‐SRP) e associados (anti‐RNP, anti PM‐Scl70, anti PM‐Scl‐100, anti‐Ku, anti‐SSA/Ro 52). Nenhum paciente tinha níveis baixos de complemento C3 e/ou C4. Quatro de cinco pacientes desenvolveram calcinose após o diagnóstico de DMJ (período médio de 46 meses, variação de 18 a 72 meses). Todos os pacientes apresentavam baixo nível sérico de vitamina D na apresentação de DMJ. Na apresentação e durante o seguimento, todos os pacientes tinham eletrocardiograma, ultrassom do abdome e do coração, raio‐X de tórax e testes de função pulmonar normais.
Apresentação clínica e seguimento a longo prazo de cinco pacientes com DMJ e lipodistrofia
| Paciente 1 | Paciente 2 | Paciente 3 | a Paciente 4 | Paciente 5 | |
|---|---|---|---|---|---|
| Idade no diagnóstico em anos, sexo | 14, F | 8, F | 8, F | 6, F | 3, M |
| Duração dos sintomas da DMJ no diagnóstico (meses) | 6 | 2 | 20 | 10 | 6 |
| Seguimento, meses (anos) | 185 (15,4) | 179 (14,9) | 70 (5,8) | 29 (2,4) | 291 (24,2) |
| Doenças associadas | Doença celíaca | Nenhuma | Constituição atópica | Nenhuma | Nenhuma |
| Curso da doença, última consulta | Apenas lipodistrofia residual, sem terapia | Apenas lipodistrofia residual, sem terapia | Crônico contínuo, TIS de baixa dose | Crônico contínuo, TIS de baixa dose | Crônico contínuo. TIS irregular |
| Lipodistrofia e calcinose | |||||
| Lipodistrofia | Generalizada | Focal | Generalizada | Parcial | Parcial |
| Distribuição | |||||
| Área zigomática, tronco, glúteo, coxas, extremidades superiores | Unilateral (esquerda) região bucinadora | Aspectos laterais da face, extremidades superiores proximais, região lombossacra, glúteo | Extremidades superiores proximais bilaterais | Cintura escapular/mãos | |
| Meses desde os sintomas da DMJ | 21 | 20 | 21 | 6 | 103 |
| Meses desde o diagnóstico da DMJ | 15 | 18 | 1 | 0 | 97 |
| Progressão | + (de parcial) | ‐ | + (de parcial) | ‐ | ‐ |
| Calcinose cutânea | ‐ | + | + | + | + |
| Meses desde o diagnóstico de DMJ | ‐ | 37 | 57 | 18 | 72 |
| DMJ no diagnóstico | |||||
| Escore MIIa | 10,4 | 12,1 | 11,2 | 12,1 | 11,3 |
| Contraturas musculares/articulares | ‐ | ‐ | + | + | + |
| Eritema facial | + | + | + | + | + |
| Sinal do heliotrópio e edema | + | + | + | + | + |
| Pápulas ou sinal de Gottron | + | + | + | + | + |
| Sinal do V | + | ‐ | ‐ | ‐ | ‐ |
| Sinal do xale | ‐ | ‐ | ‐ | ‐ | ‐ |
| IMC, hipertricose | |||||
| IMC no diagnóstico | 20,3 | 15,8 | 14,3 | 15,8 | 20,7 |
| IMC, último exame | 17,0 | 19 | 17,9 | 15,8 | 27,5 |
| Hipertricose no diagnóstico | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hipertricose, último exame | ‐ | ‐ | ‐ | + | + |
| Resultados laboratoriais e exames complementares na apresentação | |||||
| ANA, células HEp‐2 nucleoplasma, L/título | Homogêneo 160 | Homogêneo 160 | Homogêneo 320 | Homogêneo 640 | Pontilhado 80 |
| C3 (0,7‐1,73 g/L) | 0,8 | 0,9 | 1,210 | 1,1 | 0,81 |
| C4 (0,12‐,36 g/L) | 0,18 | 0,24 | 0,18 | 0,2 | 0,24 |
| CK (0‐154 U/L) | ↑ 444 | ↑ 783 | 90 | 90 | ↑ 759 |
| LDH (F <580, M <760) | ↑1691 | ↑ 859 | ↑ 1261 | ↑ 1261 | 423 |
| Vit D3 (75‐210 nmoL/L) | ↓26,4 | ↓25,8 | ↓40,5 | ↓ 36,3 | ↓11,2 |
| ENMG | Miopatia leve | Miopatia leve | Normal | Normal | Miopatia leve |
F, feminino; M, masculino; ANA, anticorpo antinuclear; DMJ, dermatomiosite juvenil; CK, creatina quinase; LDH, lactato desidrogenase; ECG, eletrocardiograma; ENMG, eletroneuromiografia; U/S, ultrassom; MII, miopatias inflamatórias idiopáticas; TIS, terapia imunossupressora.
Paciente do sexo feminino, caucasiana, de 14 anos, foi admitida com histórico de seis meses de sinais cutâneos e fraqueza muscular proximal. As características clínicas e os exames laboratoriais estão listados na tabela 1. Combinação de prednisona 60mg (1mg/kg/dia) com redução subsequente, cloroquina (CQ) 250mg/dia (posteriormente trocada para hidroxicloroquina – HCQ 200mg, 2x/dia) junto com corticosteroides tópicos, e então inibidores tópicos de calcineurina (ITC) foram iniciados. Após seis meses, metotrexato oral (MTX) 15mg semanalmente foi instituído. A lipodistrofia apareceu pela primeira vez na região bucinadora e no tronco 15 meses após o diagnóstico de DMJ (21 meses a partir dos sintomas iniciais de DMJ). Apesar da terapia, após 13 meses, o fenótipo parcial progrediu para lipodistrofia generalizada com as regiões glúteas e coxas (28 meses após o diagnóstico) e extremidades superiores (69 meses após o diagnóstico) sendo afetadas, respectivamente (fig. 1). Cinco anos após o diagnóstico, ocorreu recidiva da DMJ; seis ciclos de metilprednisolona intravenosa pulsada (IVMP) 500mg (≈ 10mg/kg/dia) por três dias e MTX oral levaram à remissão sem recidiva pelos quatro anos seguintes, mas a lipodistrofia persistiu.
Paciente 2 Paciente n° 3 com lipodistrofia generalizada progressiva de membros superiores proximais, ombros, região glútea e lombossacra, 13 meses após o diagnóstico de DMJ.")
Paciente do sexo feminino, caucasiana, de 8 anos, foi encaminhada com histórico de dois meses de eritema facial e de extremidades, juntamente com fraqueza muscular proximal e dor. Prednisona 30mg diariamente (1mg/kg/dia, posteriormente reduzida) e CQ 125mg/dia, levaram à remissão 29 meses após o diagnóstico. Atrofia subcutânea leve da região bucinadora esquerda ocorreu 18 meses após o diagnóstico (20 meses após os sintomas iniciais) e progrediu (fig. 2). Curiosamente, 37 meses após o diagnóstico e 19 meses após o início da lipodistrofia, calcinose leve foi observada na região atrófica. A dose de manutenção de CQ 125mg/dia foi administrada por nove meses no total e, em seguida, descontinuada. A paciente ficou livre de lesões e sintomas durante o período de seguimento restante, exceto por lipodistrofia com calcinose na região bucinadora esquerda.
Paciente 3 Paciente n° 2 com lipodistrofia mostrando atrofia subcutânea da região bucinadora esquerda, 48 meses após o diagnóstico de DMJ.")
Paciente do sexo feminino, caucasiana, de 8 anos, foi internada com histórico de 20 meses de eritema facial e fraqueza muscular simétrica. Um mês após o diagnóstico (21 meses após os sintomas iniciais), a lipodistrofia apareceu, primeiro nas extremidades superiores proximais. Nove ciclos de pulso de três dias (IVMP) 700mg (≈ 30mg/kg/dia), seguidos por prednisona 50mg/dia (2mg/kg/dia) com redução gradual subsequente, azatioprina 25mg, 3x/dia (3,1mg/kg/dia) e HCQ 100mg/dia (4,1mg/kg/dia) junto com ITC foram instituídos. Após nove meses, a remissão da DMJ foi alcançada, mas a lipodistrofia parcial progrediu para generalizada, pois os aspectos laterais da face, a região glútea, a região lombossacral e os ombros também foram afetados (figs. 3 e 4). Calcinose no joelho esquerdo foi observada 57 meses após o diagnóstico.
 Paciente n̊ 1 com lipodistrofia generalizada progressiva das regiões glúteas, coxas e membros superiores, 69 meses após o diagnóstico de DMJ.")

Paciente do sexo feminino, de 6 anos, foi internada com histórico de 10 meses de eritema facial, artralgia e fraqueza muscular generalizada. Seis meses após os primeiros sintomas de DMJ, atrofia bilateral do tecido adiposo subcutâneo na parte superior dos braços apareceu e permaneceu estável durante o seguimento (figs. 5 e 6). Doze ciclos de pulso de três dias (IVMP) variando de 320mg (≈ 15mg/kg/dia) a 640mg (≈ 30mg/kg/dia) no último ciclo, seguidos por prednisona 20mg/dia (0,93mg/kg/dia), mas aumentados para 40mg/dia (1,9mg/kg/dia) em um mês decorrente do agravamento da fraqueza muscular com redução subsequente, MTX 15mg SC semanalmente e HCQ 100mg/dia (4,8mg/kg/dia) junto com ITC foram instituídos. Após 12 meses, a força muscular melhorou, mas a fraqueza persistiu, especialmente nas extremidades inferiores, mas os parâmetros laboratoriais não foram notáveis. Calcinose dos joelhos e região dorsal das mãos foram observadas 18 meses após o diagnóstico de DMJ. Lipodistrofia dos braços persistiu durante o seguimento, apesar da terapia imunossupressora (TIS) crônica contínua de baixa dose (tabela 1).
 Paciente n° 4 com lipodistrofia estável mostrando atrofia bilateral na parte superior dos braços, 16 meses após o diagnóstico de DMJ.")

Paciente do sexo masculino, de 3 anos, apresentou histórico de seis meses de sinais cutâneos, mal‐estar, dor muscular e fraqueza. Curso de dois anos de TIS (prednisona oral 1‐1,5mg/kg, dois ciclos de pulso de três dias de IVMP 600mg, dois ciclos de imunoglobulina intravenosa (IVIG) e MTX 12,5‐20mg/semana) levou à melhora. A calcinose apareceu após quase seis anos no antebraço e progrediu para o cotovelo esquerdo, joelho e mão direitos (após oito anos) e glúteo (após nove anos). Atrofia do tecido adiposo subcutâneo nos músculos da cintura escapular e mãos apareceu 97 meses após o diagnóstico, sem progressão durante o seguimento. Ao longo dos 20 anos seguintes, o paciente teve múltiplos episódios de piora da fraqueza muscular e lesões cutâneas (figs. 7 e 8). A terapia de manutenção consistiu em prednisona oral com dosagens variadas, antimaláricos sistêmicos e ITC, mas o paciente não toma TIS regularmente.
 Paciente n° 5 com lipodistrofia nos músculos da cintura escapular e mãos, além de calcinose na mão direita e eritema de tronco, 290 meses após o diagnóstico de DMJ.")

Lipodistrofia é uma complicação rara da DMJ e provavelmente é subnotificada. O início da DMJ ocorre principalmente entre 4 e 10 anos, com uma proporção de 2,7:1 do sexo feminino:masculino em caucasianos.25 Resultados semelhantes foram observados no presente estudo, com início variando de 3 a 14 anos e proporção de 3,4:1 do sexo feminino:masculino. Misra et al. descreveram 35 pacientes com LDA infantil, sete dos quais tinham doença autoimune associada, mas apenas uma DMJ.11 Por outro lado, no estudo de Pope et al. com 30 crianças com LDA infantil, o diagnóstico subjacente mais frequente foi DMJ com 78%, isoladamente ou em associação a outras doenças autoimunes, como artrite reumatoide juvenil em 17%.10 A DMJ difere de outras doenças autoimunes, como o LES, que foi relatado como ocorrendo de 4 a 37 anos após o início da lipodistrofia,26 enquanto a DM geralmente precede o início da lipodistrofia.11,12 O tempo médio entre os sintomas iniciais da DMJ e o início da lipodistrofia foi de 34,2 meses (tabela 1). De um modo geral, o tempo de início é bem menor do que 3 a 4,6 anos, como relatado na literatura.10,12
Um estudo da gordura subcutânea comparou 20 pacientes com DMJ (dos quais 40% tinham lipodistrofia) com controles e mostrou que os pacientes com DMJ tinham IMC menor (a diferença entre controles e pacientes foi de 7,6±4,4kg).17 O índice de massa corporal (IMC) foi menor em comparação aos controles, mas não foi estatisticamente significante.17 Nos pacientes do presente estudo, o IMC variou de 14,3 a 20,7.
A revisão mais abrangente avaliando anormalidades clínicas e metabólicas em DMJ com lipodistrofia (28 pacientes) e comparando‐as com DMJ sem lipodistrofia foi realizada por Bingham et al.12 A análise determinou atrofia muscular, contraturas articulares, curso crônico de dermatomiosite contínua e calcinose como as características discriminatórias mais significantes ao comparar DMJ com e sem lipodistrofia. Calcinose, contraturas articulares, paniculite e atrofia muscular foram consideradas preditores para o desenvolvimento de lipodistrofia em DMJ.12 No presente relato, quatro de cinco pacientes (80%) tinham calcinose, mas em três de cinco a lipodistrofia precedeu a calcinose, enquanto em um de cinco pacientes a calcinose precedeu a lipodistrofia em dois anos.
A incidência de calcinose na DMJ é de 12% a 70%.4,5,17–19,27,28 No presente estudo, o início da calcinose após o diagnóstico de DMJ variou de 37 a 72 meses (média de 46 meses).
Relatos publicados mostraram que a IVMP é eficaz em encurtar a duração da erupção cutânea na DMJ, diminuir o comprometimento funcional e prevenir o desenvolvimento de calcinose.27,29 É bem conhecido o fato que a IVMP é inadequada para prevenir lipodistrofia e calcinose se o diagnóstico de DMJ for tardio.30 Huber et al. relataram 10 de 31 pacientes com DMJ em IVMP com calcinose versus 12 de 34 com calcinose no grupo não tratado com IVMP.5 Apesar do número limitado do presente estudo, quatro de cinco pacientes que receberam IVMP tiveram remissão parcial ou total, e enzimas musculares foram estabilizadas em todos os pacientes, mas infelizmente a lipodistrofia progrediu em dois pacientes, apesar de receberem IVMP e TIS agressiva.
Em termos de progressão, Bingham et al. declararam que de oito casos de lipodistrofia generalizada, apenas um apresentou primeiro lipodistrofia parcial, enquanto a lipodistrofia focal nunca progrediu para parcial ou generalizada.12
Bingham et al. relataram12 que nenhum dos pacientes com lipodistrofia tinha autoanticorpos específicos para miosite (MSA), o que está de acordo com os achados do presente estudo, pois todos os pacientes eram negativos para MSA. Infelizmente, não foram testados anticorpos anti‐p155, que foram relatados em 23% a 29% dos pacientes com DMJ.31 O anticorpo anti‐p155 foi encontrado com mais frequência em pacientes com DMJ com lipodistrofia em comparação com aqueles com DMJ sem lipodistrofia (seis de sete [83%] vs. 27/76 [36%]; p = 0,014), enquanto também foi mais frequente na lipodistrofia generalizada em comparação com a lipodistrofia parcial ou focal.12 Além disso, o anti‐p140 foi relatado em 23% dos pacientes com associação com calcinose.32 Em uma revisão retrospectiva de 96 pacientes com DMJ, nenhuma diferença significante em relação a idade, escores de atividade da doença ou lipodistrofia foi observada entre DMJ anti‐P155/140 positivo, DMJ com outros MSA positivos e DMJ com MSA negativo e as proporções de gordura tronco:perna foram semelhantes entre os três grupos.33 Um mecanismo potencial de lipodistrofia é a desregulação da via alternativa do complemento. A lipodistrofia tem sido associada à presença do fator nefrítico C3 do anticorpo IgG que utiliza a capacidade dos adipócitos de formar C3 convertase em suas adjacências e induz a lise mediada por complemento dos adipócitos.11 Misra et al. mostraram que 67% de seus pacientes com lipodistrofia parcial adquirida tinham baixos níveis de C3, e 83% tinham fator nefrítico C3.11 Por outro lado, Bingham et al. relataram que a maioria de seus pacientes tinha níveis normais de C3, incluindo todas as lipodistrofias parciais em pacientes com DMJ.12 Isso está de acordo com a presente análise, pois todos os pacientes do presente estudo tinham níveis normais de C3.
Relatos demonstraram que pacientes com lipodistrofia generalizada adquirida têm níveis baixos de leptina.5,34 Enquanto isso, em um estudo transversal de 59 pacientes com DMJ, a coorte com DMJ tinha níveis mais altos de leptina em comparação aos controles pareados por idade/sexo.35 Os níveis de visfatina e apelina‐12 eram mais altos em pacientes com DMJ ativa versus DMJ inativa e níveis mais baixos de adiponectina foram encontrados em pacientes com lipodistrofia associada a DMJ em comparação a pacientes com DMJ sem lipodistrofia.35 Disfunção metabólica, incluindo resistência à insulina, diabetes, hipertrigliceridemia e esteato‐hepatite não alcoólica pode ser observada. A hipertrigliceridemia foi considerada a primeira anormalidade metabólica em pacientes com DMJ, seguida por hirsutismo e lipodistrofia.17 A especificidade da hipertrigliceridemia em pacientes com DMJ e lipodistrofia é de 71% a 100% (12/18, 4/4, 17/24)7,12,17 enquanto em pacientes com DMJ sem lipodistrofia, variou de 17% (9/53)19 a 50% (10/20).17 HDL baixo, LDL e colesterol altos também estão associados com DMJ e lipodistrofia.10–12,17,35 Este é um contraste gritante, pois no presente estudo os triglicérides eram normais em todos os pacientes. O colesterol foi avaliado em todos os cinco pacientes e estava elevado apenas no paciente 3. Em segundo lugar, a resistência à insulina e o diabetes foram relatados como complicações associadas na DMJ, em percentuais mais altos nos pacientes com lipodistrofia, especialmente nas formas generalizada e parcial.5,7,12 Com base em 20 pacientes com DMJ (oito dos quais tinham lipodistrofia), um estudo mostrou que nenhum paciente tinha teste oral de tolerância à glicose (TOTG) anormal.17 Nenhum dos pacientes do presente estudo tinha níveis elevados de glicemia de jejum, apesar da terapia com corticosteroides. Há debate se as anormalidades metabólicas no estado lipídico e a hiperinsulinemia são causadas pelo tratamento com corticosteroides ou pela DMJ e/ou lipodistrofia.11 Ilowite et al. mostraram diferença nos padrões característicos de dislipoproteinemia decorrente de corticosteroides em comparação com a atividade da doença em pacientes pediátricos com lúpus. Os corticosteroides resultaram em HDL, VLDL e triglicerídeos elevados, enquanto a atividade da doença causou HDL baixo, VLDL e triglicérides elevados.36 Portanto, HDL baixo e triglicérides elevados indicaram que a dislipoproteinemia estava relacionada à doença.7,12
Em termos de biomarcadores validados para atividade de DMJ, neopterina, CXCL11 e galectina‐9 foram considerados prospectivos.37 Além disso, IFN tipo I elevado foi correlacionado com atividade de doença de pele e o escore do gene regulado por interferon foi significantemente maior em DMJ do que em controles, correlacionando‐se moderadamente com atividade de DMJ e fortemente com atividade cutânea em pacientes anti‐TIF1 positivos.38
Complicações metabólicas em outras formas de lipodistrofia demonstraram responder a tiazolidinedionas (troglitazona 200–600mg diariamente), causando diminuição dos níveis de hemoglobina A1c, níveis de triglicérides, níveis de ácidos graxos livres e aumento de gordura corporal em média de 2,4 pontos percentuais.39 Lebastchi et al. relataram três casos de lipodistrofia associada a doenças autoimunes (uma criança tinha DMJ), mostrando que o tratamento de quatro a seis anos com metreleptina (0,02–0,04mg/kg SC) reduziu a resistência à insulina e a hipertrigliceridemia sem alterar ou piorar a doença autoimune.34
Todos os pacientes do presente estudo apresentavam baixo nível sérico de vitamina D na apresentação da DMJ (tabela 1). O papel essencial da vitamina D na homeostase óssea é bem conhecido, mas a vitamina D tem múltiplos efeitos imunomoduladores, incluindo a regulação da resposta imune. Anteriormente, foi demonstrado que a deficiência de vitamina D está associada à atividade da doença em pacientes com DMJ.40 É importante que crianças e adolescentes suplementem a vitamina D de acordo com as recomendações atuais. Mais estudos bem elaborados são necessários para determinar a significância potencial dos baixos níveis séricos de vitamina D que foram encontrados em todos os pacientes do presente estudo com lipodistrofia associada à DMJ.
ConclusãoA lipodistrofia é complicação rara, significante e provavelmente subnotificada da DMJ. Durante o período de 34 anos, cinco de 22 (23%) pacientes com DMJ desenvolveram lipodistrofia. Não foram encontradas complicações metabólicas. Todos os cinco pacientes tiveram atraso significante no diagnóstico da DMJ. Apesar da TIS padrão subsequente, dois de cinco pacientes progrediram para lipodistrofia generalizada e quatro de cinco pacientes desenvolveram calcinose. A implementação precoce de TIS intensiva é necessária para reduzir o risco de lipodistrofia, como uma complicação crônica da DMJ. Mais estudos prospectivos usando medidas de desfecho validadas são necessários para uma terapia mais precisa e evolução a longo prazo da lipodistrofia associada à DMJ.
Suporte financeiroNenhum.
Contribuição dos autoresIgor Kapetanović: Aprovação da versão final do manuscrito; revisão crítica da literatura; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; elaboração e redação do manuscrito; análise estatística; concepção e planejamento do estudo; revisão crítica do manuscrito.
Mirjana Gajić‐Veljić: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Branka Bonači‐Nikolić: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito.
Miloš Nikolić: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Conflito de interessesNenhum.
MN, BBN e MGV receberam suporte do Ministério da Educação, Ciência e Desenvolvimento Tecnológico da República da Sérvia, Subvenção n° 100200.
Como citar este artigo: Kapetanović I, Gajić‐Veljić M, Bonači‐Nikolić B, Nikolić M. Long‐term outcome of juvenile dermatomyositis associated with lipodystrophy: experience of a university hospital. An Bras Dermatol. 2025;100:501130.
Trabalho realizado no University Clinical Center of Serbia e University of Belgrade, Belgrado, Sérvia.
