


Recently, the World Health Organization published the revised 4th edition of its classification of tumors of hematopoietic and lymphoid tissues. The present paper is a concise comparative review of the main primary cutaneous T-cell hematopoietic tumors, with emphasis on their immunohistochemical profiles.
The World Health Organization’s recently published revised 4th edition of the classification of tumors of the hematopoietic and lymphoid tissues reflects the advances in the molecular and pathological understanding of these lesions, establishing new criteria for primary cutaneous lymphomas. 1,2
Cutaneous T-cell lymphomas are represented mainly by mycosis fungoides (MF) and CD30+ T-cell lymphoproliferative disorders.1,3,4 Thus, the present paper offers a brief explanation of those entities, establishing a comparative immunohistochemical panel using the Portuguese language nomenclature.5
Mycosis FungoidesRegarding the immunohistochemical profile, we discuss whether there is loss of CD7 expression in MF, or whether MF is a neoplasm of CD7-negative helper T lymphocytes.3 Another point is CD8 positivity of neoplastic cells, which is more common in the pediatric population.1 In addition, the presence of CD30-positive cells in plaque stage does not seem to have a prognostic significance, unlike cases with transformation to large cell lymphoma when their positivity is associated with better survival.3
Primary Cutaneous CD30+ T-Cell Lymphopro-Liferative DisordersThis category is the second most frequent group of cutaneous T-cell lymphomas, corresponding to approximately 30% of the cases.1,4,6
Lymphomatoid papulosis (LyP)In addition to the previously described variants A, B and C, other forms have been introduced. Type D is characterized by epidermotropic CD8+ and CD30+ T-cell lymphocytes, differentiated from type B by the immunohistochemical profile. Type E is defined as small- to medium-sized pleomorphic T cell neoplasia with CD8+ and CD30+ immunophenotype and angiocentric and angiodestructive infiltration.1,7 Positivity for CD567 is rarely observed. LyP with 6p25.3 rearrangement corresponds to less than 5% of the cases that are characterized by rearrangement involving DUSP22-IRF4 in the 6p25.3 locus, which presents a histopathological picture resembling that of MF in transformation to large cell lymphoma (Table 1).1
Classification of lymphomatoid papulosis according to the revised 4th edition of WHO’s classification of hematopoietic tumors1
| Type | Frequency | Histology | Immunohistochemistry |
|---|---|---|---|
| A | >80% | Sparse large and atypical neoplastic cells amid mixed inflammatory infiltrate | CD4+, CD8- |
| B* | <5% | Epidermotropic Infiltrate of small lymphocytes | CD4+, CD8- |
| C | ~10% | Cohesive infiltrate of large and atypical cells among sparse inflammatory cells | CD4+, CD8- |
| D | <5% | Infiltration of small and medium atypical epidermotropic cells. | CD4-, CD8+ |
| E | <5% | Angiocentric and angiodestructive pattern | CD4-, CD8+ |
| Rearrangement DUSP22-IRF4 | <5% | Small and epidermotropic lymphocytes associated with large neoplastic cells in the dermis (similar to transforming MF) | CD4-, CD8+ ou CD4-, CD8- |
LyP type B can be negative for CD30.
Source: Swerdlow, et al., 2017.1
This diagnosis always requires the exclusion of any evidence or history of MF, which may present a CD30+ or CD30- tumor evolution. Clinically, C-ALCL is a localized, ulcerated lesion appearing in elderly people, on the face, trunk or gluteus, with a 5-year survival rate above 90%.8 The vast majority of C-ALCL cases are negative for anaplastic lymphoma kinase 1 (ALK) epithelial membrane antigen.1,4 ALK protein positivity may occur more frequently in children and may correspond to a higher probability of progression to the systemic form.8 There may be spontaneous regression in 20 to 42% of the cases, with relapse occurring in half of them.7,8
Subcutaneous Panniculitis-Like T-Cell LymphomaA rare CD8+ cytotoxic αβ T-cell lymphoma, with involvement restricted to subcutaneous tissue,3 is associated with autoimmune diseases in 20% of the cases. It has a good prognosis, with neoplastic cells surrounding individual adipocytes. Positive gamma-delta cases should be excluded from this category.1,3,4,6 The absence of plasma cells and clusters of CD123-positive plasmacytoid dendritic cells aid in the differentiation of lupus panniculitis.7
Primary Cutaneous Gamma Delta T-Cell LymphomaIt is defined as a clonal proliferation of mature and activated gamma-delta T cells expressing cytotoxic markers. Clinically, it is characterized by infiltrated plaques on legs, trunk and arms, usually in elderly people.9 In 60% of cases, erosion or necrosis occurs. Histologically, the disease is variable and can affect the epidermis, dermis and subcutaneous tissues by medium to large cell infiltrates with frequent angioinvasion and necrosis. LyP and MF cases can express gamma-delta receptors; however, these cases should continue to be diagnosed as LyP and MF. It has a poor prognosis.1,4,6 and subcutaneous involvement is an indicator of worse prognosis.1,9 The estimated 5-year survival is 19.9%.9
Primary Cutaneous CD8+ Aggressive Epidermotropic Cytotoxic T-Cell LymphomaProvisional entity characterized by epidermotropic CD8+ T-cell lymphoma that demonstrates aggressive clinical behavior. The cells vary in size from small to large. Epidermotropism can be intense or even localized. Ulceration, necrosis, adnexal destruction and blister formation are common. When there is CD30 expression, the histopathological and immunohistochemical profile is indistinguishable from LyP type D; differentiation is based on the clinical history.1 It presents an estimated 5-year survival of 0%,3 which is not related to cell morphology (small versus large cells) nor to the presence of localized or diffuse disease.1
Primary Cutaneous Acral CD8+ T-Cell LymphomaA rare clonal cutaneous infiltration of CD8-positive medium-sized cytotoxic T lymphocytes. It involves the acral skin, with ears being the most common site. It usually does not affect the epidermis, presents neither mitotic figures nor necrosis, and has a good prognosis.1,3
Primary Cutaneous CD4+ Small/Medium-Sized T-Cell Lymphoproliferative DisorderPreviously called “primary cutaneous CD4+ small/medium T-cell lymphoma”, it was renamed “lymphoproliferative disorder” because it is a localized neoplasm with a certain resemblance to clonal proliferations secondary to medication use. Characterized by small- to medium-sized atypical CD4+ T cells, it usually appears as a single nodular lesion or plaque on the head and neck. This diagnosis requires that the hypothesis of MF be excluded, emphasizing that a small proportion of large and pleomorphic cells (<30%) can be identified. The prognosis is good, depending on whether it is a type of lymphoma, a precursor lesion, a totally benign lesion or a pseudolymphoma.1,3
Hydroa Vacciniforme-Like Lymphoproliferative DisorderHydroa vacciniforme-like lymphoma has started to be considered a lymphoproliferative disorder due to its clinical presentation and its relation to active Epstein-Barr virus infection. It is a chronic lesion occurring in childhood and is associated with a risk of developing systemic lymphoma. It should be classified into two types: the classic self-limited type, characterized by vesicles in sun-exposed areas and a benign course; and the severe type, with extensive cutaneous involvement and systemic manifestations such
as fever, hepatomegaly and lymphocytosis in peripheral blood.3 There is no morphological or clinical basis capable of predicting biological behavior.1
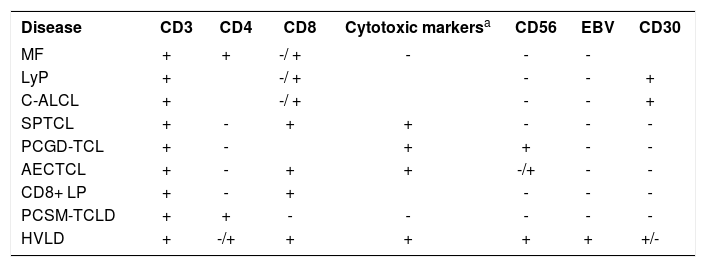
ConclusionIn summary, based on the abovementioned book, we suggest a practical immunohistochemical approach according to table 2. However, it is emphasized that many cases escape the typical immunophenotype and must be interpreted with other clinical and histopathological data. □
Differential diagnosis of cutaneous T cell neoplasms
| Disease | CD3 | CD4 | CD8 | Cytotoxic markersa | CD56 | EBV | CD30 |
|---|---|---|---|---|---|---|---|
| MF | + | + | -/ + | - | - | - | |
| LyP | + | -/ + | - | - | + | ||
| C-ALCL | + | -/ + | - | - | + | ||
| SPTCL | + | - | + | + | - | - | - |
| PCGD-TCL | + | - | + | + | - | - | |
| AECTCL | + | - | + | + | -/+ | - | - |
| CD8+ LP | + | - | + | - | - | - | |
| PCSM-TCLD | + | + | - | - | - | - | - |
| HVLD | + | -/+ | + | + | + | + | +/- |
= Granule-associated cytotoxic proteins: TIA1, granzyme B, perforin; b = TIA1 can present positive expression in dot pattern; MF: mycosis fungoides; LyP: lymphomatoid papulosis; C-ALCL: Primary cutaneous anaplastic large cell lymphoma; SPTCL: Subcutaneous panniculitis-like T-cell lymphoma; PCGD-TCL: Primary cutaneous gamma delta T-cell lymphoma; AECTCL: Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma; CD8+ LP: Primary cutaneous acral CD8+ T-cell lymphoma; PCSM-TCLD: Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder; HVLD: Hydroa vacciniforme-like lymphoproliferative disorder
Financial support: None.
Conflict of interest: None.
