





Autoimmune progesterone dermatitis is an uncommon, poorly recognized and under-diagnosed catamenial dermatosis associated with hypersensitivity reactions to progestagens. Most cases manifest as urticaria, eczema or erythema multiforme-like. A 26-year-old woman developed violaceous plaques on the groin and abdomen, 4 days after a spontaneous abortion resolved with uterine curettage. The lesions recurred once monthly at the same sites, mimicking a fixed drug eruption. Although the histopathology was compatible with fixed drug eruption, positive intradermal testing and symptomatic improvement after using oral contraceptive pills gave us a clue to the diagnosis.
Autoimmune progesterone dermatitis (APD) is a rare form of hypersensitivity (HS) to progesterone (PG). It is characterized by skin lesions that occur during the luteal phase of the menstrual cycle, coinciding with peak levels of endogenous PG.
Case ReportA 26-year-old female patient presented with a 5-month history of recurring skin lesions associated with itching and burning sensations. Symptoms first began 4 days after uterine curettage following a missed abortion. The patient’s history was otherwise clear, except for irregular use of combined oral contraceptive pills (OCP) (levonorgestrel; ethinylestradiol). The lesions recurred once monthly at the same sites, beginning one day prior to menstruation and resolving 3 days after the end of the menstrual cycle, leaving residual hyperpigmentation. They were initially treated with oral antihistamines and clobetasol cream, with no response. Physical examination revealed sharply demarcated violaceous plaques located at the armpits, groin, labia majora, intergluteal cleft, and abdomen (Figures 1 and 2). The patient’s complete blood count, erythrocyte sedimentation rate, blood biochemistry, and immunological and hormone panels were all within normal ranges. Histopathological analysis of skin lesions was compatible with fixed drug eruption (FDE) (Figure 3). In light of suspicion of APD due to catamenial recurrence with normal laboratory results, an intradermal test was performed with 0.5ml progesterone (50mg/ml aqueous solution, undiluted); this gave a positive result in 3 minutes, manifesting with erythema, edema, and pruritus, which lasted approximately 6 hours (Figure 4). The patient was diagnosed with generalized FDE-like APD, and prescribed OCP with dienogest and ethinylestradiol as initial treatment to suppress ovulation. Currently, the patient has been monitored for 5 months, with no recurrence of skin complaints.


")

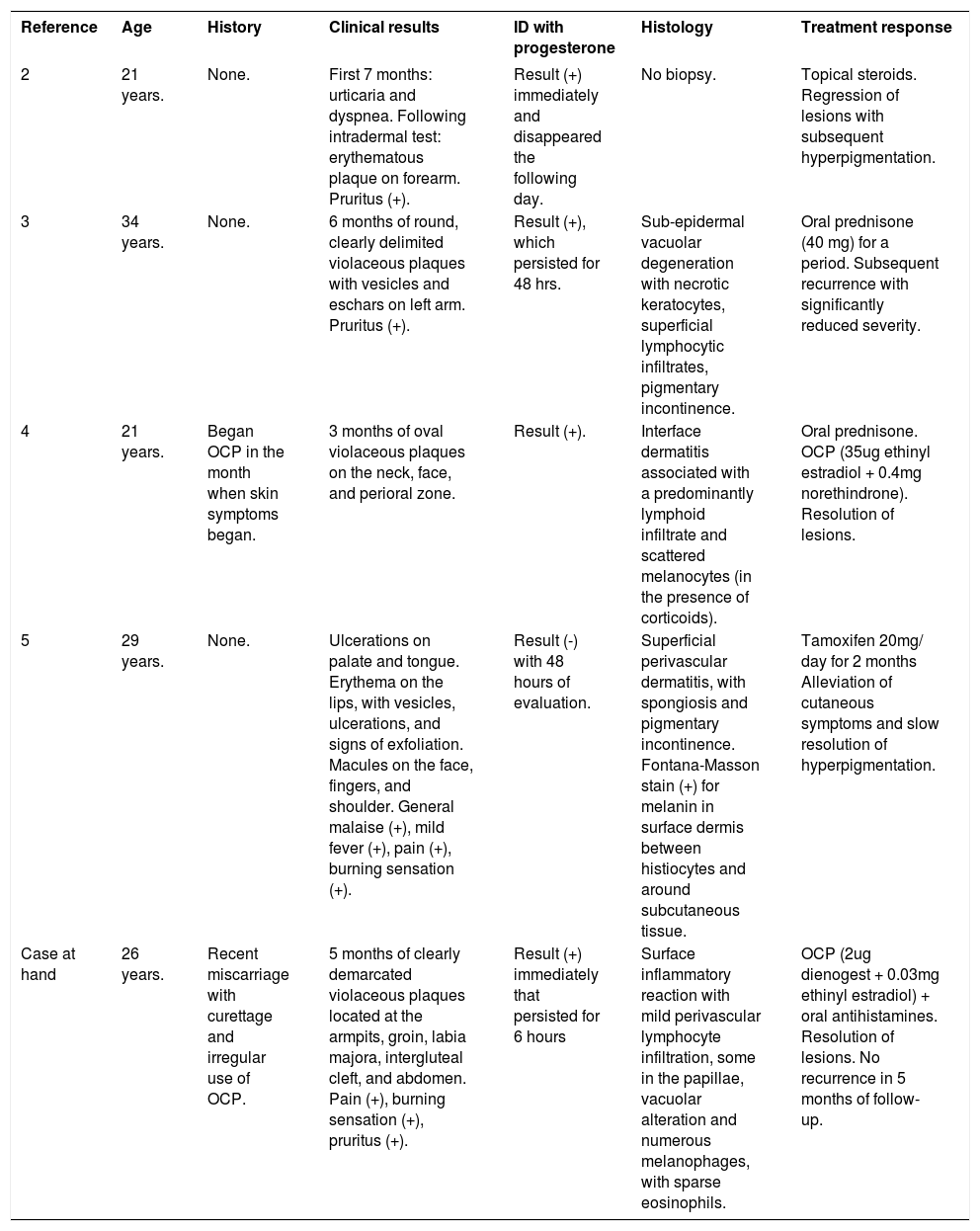
Published literature of APD describes at least 8 forms of skin manifestations. Most frequent presentations are: urticaria, eczema, and erythema multiforme-like lesions, although a recent report showed that half of cases were found to manifest as urticaria, vesiculobullous eruptions, erythema multiforme, eczema, and maculopapular rashes.1 It is noteworthy that to date, only 4 cases of FDE-like APD have been reported (Table 1).2-5
Reported cases of autoimmune progesterone dermatitis manifesting as fixed drug eruption
| Reference | Age | History | Clinical results | ID with progesterone | Histology | Treatment response |
|---|---|---|---|---|---|---|
| 2 | 21 years. | None. | First 7 months: urticaria and dyspnea. Following intradermal test: erythematous plaque on forearm. Pruritus (+). | Result (+) immediately and disappeared the following day. | No biopsy. | Topical steroids. Regression of lesions with subsequent hyperpigmentation. |
| 3 | 34 years. | None. | 6 months of round, clearly delimited violaceous plaques with vesicles and eschars on left arm. Pruritus (+). | Result (+), which persisted for 48 hrs. | Sub-epidermal vacuolar degeneration with necrotic keratocytes, superficial lymphocytic infiltrates, pigmentary incontinence. | Oral prednisone (40 mg) for a period. Subsequent recurrence with significantly reduced severity. |
| 4 | 21 years. | Began OCP in the month when skin symptoms began. | 3 months of oval violaceous plaques on the neck, face, and perioral zone. | Result (+). | Interface dermatitis associated with a predominantly lymphoid infiltrate and scattered melanocytes (in the presence of corticoids). | Oral prednisone. OCP (35ug ethinyl estradiol + 0.4mg norethindrone). Resolution of lesions. |
| 5 | 29 years. | None. | Ulcerations on palate and tongue. Erythema on the lips, with vesicles, ulcerations, and signs of exfoliation. Macules on the face, fingers, and shoulder. General malaise (+), mild fever (+), pain (+), burning sensation (+). | Result (-) with 48 hours of evaluation. | Superficial perivascular dermatitis, with spongiosis and pigmentary incontinence. Fontana-Masson stain (+) for melanin in surface dermis between histiocytes and around subcutaneous tissue. | Tamoxifen 20mg/ day for 2 months Alleviation of cutaneous symptoms and slow resolution of hyperpigmentation. |
| Case at hand | 26 years. | Recent miscarriage with curettage and irregular use of OCP. | 5 months of clearly demarcated violaceous plaques located at the armpits, groin, labia majora, intergluteal cleft, and abdomen. Pain (+), burning sensation (+), pruritus (+). | Result (+) immediately that persisted for 6 hours | Surface inflammatory reaction with mild perivascular lymphocyte infiltration, some in the papillae, vacuolar alteration and numerous melanophages, with sparse eosinophils. | OCP (2ug dienogest + 0.03mg ethinyl estradiol) + oral antihistamines. Resolution of lesions. No recurrence in 5 months of follow-up. |
Abbreviations: ID, intradermal test; OCP, oral contraceptive pills.
Our patient presented with violaceous plaques always at the same sites -findings suggestive of FDE- but the periodic nature of the pathology, its relationship with menstruation, and the patient not having taken any medications led to a suspicion of APD. An analysis of the cases of FDE-like APD described in the literature found that from a histological perspective, 3 of them were compatible with adverse drug reactions (ADR).3-5
The diagnostic criteria for APD proposed by Warin et al6 are: (1) skin lesions related to menstrual cycle; (2) symptomatic improvement after inhibiting PG secretion by suppressing ovulation; (3) positive response to intradermal testing with PG. However, to date there is no standardization regarding the dose, environmental conditions, or the technique to use when performing this test on patients suspected to have APD. Therefore, the test is not a reliable predictor of the phenomenon of HS to PG.7 In a series of 24 cases of HS to PG, only 50% of patients showed a positive result to this test.8 Histology is of little help, as no specific findings have been described. It is probable that the presentation and pathogenic mechanisms depend on the type of skin manifestation affecting each patient.
The causes of APD are unknown. It has been proposed that it is an HS reaction to PG, and that it could be part of a broader syndrome of HS to some or all sex steroids (SS), including estrogens. This intolerance is believed not to be uncommon and is thought to manifest in different ways including dermatitis, erythema multiforme, dysmenorrhea, premenstrual syndrome and mastalgia.9
The pathogenic mechanisms of APD may be diverse and they are probably similar to HS reactions to drugs-with the participation of IgE antibodies, T cells, or proinflammatory cytokines.9 A significant percentage of patients with APD show immediate or delayed positive reactions to intradermal tests with PG, suggesting that type I and IV HS reactions may be involved. Given that immunological mechanisms have not been confirmed in all patients, Foer et al8 recently suggested that this pathology should be renamed as HS to PG instead of APD, as this feature is indeed shared by all patients. They also recommend a new classification, in which patients exhibit 2 phenotypes: 1) those in whom it is triggered by endogenous PG (situations such as menstruation and pregnancy) with symptoms emerging during the luteal phase, and 2) those who had previously tolerated menstruation and in some cases even pregnancy, but symptoms emerged following exposure to exogenous PG. This latter group has been considered to have a mixed phenotype, as following sensitization they suffer exacerbation from endogenous triggers.8
It has been shown that 14% of cases of ADP are triggered by pregnancy, with symptoms usually beginning during interpartum or postpartum.1 The alleviation of ADP experienced by some patients during pregnancy may relate to a reduction in natural cell-mediated immunity, allowing the fetus to be tolerated. Nonetheless, case studies show that not all patients respond in the same way to HS to PG. Indeed, a case of miscarriage potentially caused by HS to PG has been reported.10 Furthermore, it has been seen that implantation and success of pregnancy depend on suitable immune responses. Cases of recurring miscarriages have been associated with an excess of Th1 proinflammatory cytokines compared to levels of type Th2/3.9 Although miscarriages are caused by multiple factors, the time course of the miscarriage and the emergence of ADP in our patient suggests a possible role of autoimmunity and a potential shared etiopathology.
From a physiopathological perspective, ovulation must be suppressed to reduce endogenous PG production. Complete resolution of lesions has been achieved with the use of conjugated equine estrogens, ethinylestradiol, OCP combined with progestins, gonadotropin-releasing hormone analogs and tamoxifen.1 Variable results have been achieved with the use of systemic corticoids and antihistamines.4 Definitive therapy is hysterectomy with salpin-go-oophorectomy, which should be reserved for severe cases, and patients with no future desire to have children.1,4 Desensitization with increasing doses of PG (intramuscular, oral or intravaginal) is also reported and has shown success in controlling symptoms and eventually achieving fertility.1,4-5,8
Treatment must be selected based on the patient’s profile and preferences.
Financial support: None.
Conflict of interest: None.
