




O carcinoma de células de Merkel é câncer de pele raro com diferenciação neuroendócrina. Os fatores de risco incluem exposição solar, idade avançada, imunossupressão (como pacientes transplantados, pacientes com neoplasias linfoproliferativas ou portadores do vírus HIV) e infecção pelo poliomavírus de células de Merkel. Clinicamente, o carcinoma de células de Merkel aparece como placa ou nódulo cutâneo ou subcutâneo, mas o diagnóstico desse tumor raramente é feito clinicamente. Assim, a histopatologia e a imuno‐histoquímica costumam ser necessárias. Tumores primários sem evidência de metástases são tratados com excisão cirúrgica completa com margens cirúrgicas apropriadas. A presença de metástase oculta em linfonodo é frequente, e a biópsia do linfonodo sentinela deve ser realizada. A radioterapia adjuvante pós‐operatória aumenta o controle local do tumor. Recentemente, agentes que bloqueiam a via PD‐1/PD‐L1 mostraram regressões tumorais objetivas e duráveis em pacientes com neoplasias sólidas avançadas. O primeiro anticorpo anti‐PD‐L1 usado em pacientes com carcinoma de células de Merkel foi o avelumabe, mas pembrolizumabe e nivolumabe também demonstraram eficácia. Neste artigo, o estado atual do conhecimento da epidemiologia, diagnóstico, estadiamento e as novas estratégias de tratamento sistêmico do carcinoma de células de Merkel são descritos.
O carcinoma de células de Merkel (CCM) é um tipo incomum e agressivo de câncer de pele, com origem neuroendócrina,1 descrito pela primeira vez em 1972 por Toker como um carcinoma trabecular da pele.2 Desde então, o entendimento sobre sua fisiopatologia, resultados e manejo tem crescido, principalmente após as imunoterapias. Também conhecido como carcinoma neuroendócrino cutâneo primário, o CCM compartilha características com as células de Merkel da pele, justificando sua nomenclatura.3
EpidemiologiaA taxa de incidência de CCM varia de acordo com as distintas regiões do mundo. Na União Europeia, entre 1995 e 2002, sua taxa de incidência anual foi de 0,13/100.000 habitantes, e mostrou‐se maior nos grupos com 65 anos ou mais.4 Nos EUA, a taxa de incidência foi de 0,79/100.000 habitantes em 2011.5 Em 2013, Paulson et al. relataram que o número absoluto de CCM nos EUA foi de 2.488 casos, o que corresponde a uma taxa de incidência semelhante a 0,7/100.000 pessoas‐ano.6
Maior incidência de CCM foi observada em estudo australiano, com taxa de 1,6/100.000 habitantes no estado de Queensland entre 2006 e 2010, mais frequente no sexo masculino (2,5/100.000) do que no feminino (0,9/100.000) e com expectativa de vida média de 75,5 anos para homens e 78 anos para mulheres no momento do diagnóstico.7
Embora rara, a incidência de CCM cresceu drasticamente.8 Em 2018, Paulson et al. relataram que a quantidade de casos aumentou 95% entre 2000 e 2013 nos EUA, em comparação com um aumento de 57% para melanoma e 15% para todos os demais tumores sólidos.6 Na Austrália, a incidência geral de CCM aumentou em média 2,6% ao ano entre 1993 e 2010 (95%IC 1,1%‐4,2%).7 O CCM é frequentemente subdiagnosticado, e acredita‐se que parte de sua incidência aumentada seja decorrente de patologistas mais bem treinados e também do desenvolvimento de biomarcadores que melhoraram a detecção da doença.6
No que diz respeito à epidemiologia do CCM no Brasil, Melo et al. publicaram um estudo com dados coletados dos Registros de Câncer de Base Populacional (2000‐2015) e de Base Hospitalar (2000‐2017). Foram analisados 881 pacientes com a doença; a maioria era do sexo feminino (51,2%), com idade maior que 60 anos (82,2%), de cor branca (67,6%) e com diagnóstico predominantemente nos estádios III ou IV (50,5%). Além disso, constatou‐se que as taxas médias de incidência padronizadas por idade aumentaram significantemente em homens entre os anos de 2000 (0,31/1.000.000) e 2015 (1,21/1.000.000), com variação percentual anual de 9,4 (95%IC 4,7%‐14,4%; p<0,001). Nas mulheres, as taxas de incidência não aumentaram significantemente no período.9
Carneiro et al. avaliaram retrospectivamente 32 pacientes atendidos no Instituto Nacional de Câncer com CCM entre 2002 e 2012, e a média de idade ao diagnóstico foi de 72 anos. No entanto, diferentemente dos dados internacionais, a maioria dos pacientes era do sexo feminino (69%).10
Fatores de riscoVários estudos sugerem que múltiplos fatores podem contribuir para o desenvolvimento do CCM.7,8,11–13 A exposição solar é importante fator de risco,13,14 corroborado pela maior incidência de CCM em regiões com maior índice de radiação ultravioleta7,8 e pela maior tendência a ocorrer em áreas fotoexpostas, como região de cabeça e pescoço.15 Lunder et al. também mostraram maior ocorrência de CCM em pacientes com história de uso de psoralenos e exposição da pele à radiação ultravioleta de ondas longas.16 O CCM é mais frequente em pacientes caucasianos em comparação com outras etnias.8
A imunossupressão também é fator de risco;13 portanto, o CCM é mais comum em pacientes transplantados, com diagnóstico de neoplasias linfoproliferativas (como a leucemia linfocítica crônica) ou em portadores do vírus HIV.8,14
Além disso, idade avançada também é considerada fator de risco.13,15 Em uma série dos EUA, 90% de todos os pacientes com CCM estavam acima de 50 anos, 76% acima de 65 anos e 49% acima de 75 anos.8,15
Em 2008, Feng et al. descreveram pela primeira vez a associação entre um novo poliomavírus e o CCM, identificando o DNA do vírus em 8 de 10 tumores de Merkel, sugerindo que a infecção viral poderia ser um evento precoce na patogênese.11
Poliomavírus associado ao carcinoma de células de MerkelA descoberta do poliomavírus associado ao CCM, em 2008, revolucionou os conhecimentos sobre a patogênese desse tumor, então denominado Merkel cell polyomavirus (MCPyV).11 A associação foi identificada usando‐se técnica de subtração de transcriptoma digital, que é um método de bioinformática para detectar a presença de novos transcritos de patógenos por meio da remoção computacional das sequências do hospedeiro. Então, nesse caso, sequências humanas conhecidas foram filtradas para identificar potenciais transcritos virais.3
Os casos de infecção subclínica por MCPyV aumentam de acordo com a senescência, atingindo prevalência de 60% a 80% em adultos. No entanto, isso pode diferir significantemente em outras regiões geográficas, como a Austrália, onde foi relatada associação muito menor com infecção viral, em torno de 25%.17 A pele constitui o maior sítio da infecção viral, apesar de o vírus ter sido detectado também no sangue periférico e em outros órgãos. A infecção pelo MCPyV parece ser assintomática.3
A família Polyomaviridae, a qual o MCPyV pertence, é composta por pequenos vírus de DNA dupla‐hélice. Ela inclui outros poliomavírus associados com infecção cutânea em humanos (poliomavírus da Tricodisplasia Spinulosa, poliomavírus humano 6 e poliomavírus humano 7) ou doenças em outros órgãos e sistemas (como o vírus JC, um poliomavírus humano ubíquo que causa doenças do sistema nervoso central em pacientes imunocomprometidos, incluindo leucoencefalopatia multifocal progressiva, neuropatia de células granulares e encefalopatia pelo vírus JC). Até a presente data, o MCPyV é o único oncovírus humano da família Polyomaviridae, mas o motivo desse status distinto ainda não é conhecido.3
O tipo específico de célula hospedeira para a infecção por esse poliomavírus ainda permanece incerto. Acredita‐se que as células de Merkel não sejam suficientemente numerosas para explicar a carga viral normalmente detectada na pele, e os monócitos no sangue periférico têm sido apontados como possível reservatório de células infectadas.3
O genoma viral epissomal apresenta regiões precoces e tardias. As primeiras têm genes que codificam proteínas responsáveis por coordenar a replicação viral, enquanto as regiões tardias estão ligadas às proteínas do capsídeo viral. Parece que a integração do DNA do MCPyV no genoma do hospedeiro ocorre logo após a infecção.3,18
Imuno‐histoquímica, PCR, hibridização in situ de DNA ou tecnologias de sequenciamento de nova geração são métodos disponíveis para detectar MCPyV em tumores, mas esses testes variam muito em sensibilidade e especificidade. O desenvolvimento de um anticorpo monoclonal usado para detecção específica do antígeno T grande (LT) por meio da imuno‐histoquímica ‐ exclusivo para o MCPyV (o clone CM2B4) ‐ possibilitou a detecção e visualização de MCPyV in situ. Esse método está disponível comercialmente e tem aproximadamente 88% de sensibilidade e 94% de especificidade.3 A figura 1 demonstra exemplos de resultados de imuno‐histoquímica positivo e negativo para o poliomavírus de células de Merkel realizada utilizando o anticorpo anti‐MCPyV large T‐antigen (CM2B4).
 Lâmina de imuno‐histoquímica de paciente apresentando resultado positivo para MCPyV (aumento de 400×). (B) Lâmina de imuno‐histoquímica de paciente apresentando resultado negativo para MCPyV (aumento de 400×).")
Imuno‐histoquímica para MCPyV realizada em lâminas de carcinoma de células de Merkel. (A) Lâmina de imuno‐histoquímica de paciente apresentando resultado positivo para MCPyV (aumento de 400×). (B) Lâmina de imuno‐histoquímica de paciente apresentando resultado negativo para MCPyV (aumento de 400×).
Clinicamente, o CCM pode se apresentar como nódulo cutâneo ou subcutâneo, e até mesmo ter aspecto cístico. A cor pode variar entre vermelho, rosa, azul, violeta ou cor da pele. Inicialmente, as lesões são geralmente indolores e solitárias, mas também podem ulcerar ou ser circundadas por lesões satélites. No diagnóstico, as dimensões podem variar em tamanho, mas costumam ser menores que 20 mm, e a maioria dos casos apresenta crescimento rápido do tumor em poucos meses (fig. 2).15
 Ao diagnóstico, lesão tumoral com aspecto cupuliforme, com eritema e discreta descamação. (B) Evolução do tumor com crescimento e ulceração em toda a lesão. Fonte: Cortesia da Dra. Gabriella Campos‐do‐Carmo. O paciente consentiu com o uso das imagens para fins educacionais.")
Carcinoma de células de Merkel localizado na coxa esquerda de paciente do sexo masculino. (A) Ao diagnóstico, lesão tumoral com aspecto cupuliforme, com eritema e discreta descamação. (B) Evolução do tumor com crescimento e ulceração em toda a lesão.
Fonte: Cortesia da Dra. Gabriella Campos‐do‐Carmo. O paciente consentiu com o uso das imagens para fins educacionais.
As lesões geralmente aparecem em áreas fotoexpostas, enquanto 19% aparecem nas nádegas ou em áreas minimamente expostas ao sol. Os sítios anatômicos primários mais frequentes são cabeça e pescoço (29%), seguidos por membros inferiores (24%) e membros superiores (21%).15
Heath et al., em 2008, propuseram a regra mnemônica “AEIOU” (Assintomatic/lack of tenderness, Expandig rapidly, Immune suppression, Older than 50 years, and Ultraviolet exposed site) a partir da análise de 195 casos, na tentativa de auxiliar o diagnóstico. Nessa coorte, 89% dos pacientes com diagnóstico de CCM preencheram três ou mais critérios, 52% dos pacientes preencheram quatro ou mais critérios e 7% dos pacientes preencheram todos os cinco critérios.15
No momento do diagnóstico, até 37% dos pacientes apresentam doença nodal e 6%‐12% apresentam‐se com doença metastática.19 Cerca de 15% dos pacientes têm um linfonodo acometido pela doença sem identificar lesão cutânea, provavelmente refletindo a regressão do sítio primário.3,15
As metástases a distância aparecem com mais frequência em linfonodos não regionais, pele, ossos, pulmões e pleura ou fígado e, menos comumente, pâncreas, glândulas suprarrenais, cérebro, rins, tecido subcutâneo ou músculo. Os locais raros de metástases incluem mama, trato gastrintestinal, testículos, coração, retroperitônio e peritônio.14
DiagnósticoEm decorrência da falta de características e de sintomas específicos, o diagnóstico torna‐se um desafio. Assim, o exame histopatológico por patologista experiente e a coloração com marcadores imuno‐histoquímicos específicos são essenciais nesse processo.3
A organização americana National Comprehensive Cancer Network (NCCN) propôs algoritmos específicos para o diagnóstico de CCM, que incluem as seguintes estratégias: 1) exame completo de pele e linfonodos; 2) biópsia para análise histopatológica e imuno‐histoquímica; 3) biópsia de linfonodo sentinela (BLS) para pacientes sem linfonodos clinicamente positivos, precedendo a excisão, se possível; 4) aspiração por agulha fina ou biópsia linfonodal para pacientes com linfonodos clinicamente positivos; considerar biópsia aberta; 5) exames de imagem para estadiamento, conforme indicação clínica.14
A dermatoscopia é um exame complementar que pode ser útil em alguns casos, mas ainda faltam descrições dermatoscópicas mais completas na literatura. O CCM geralmente exibe uma variedade de padrões vasculares dermatoscópicos, mais comumente áreas/glóbulos vermelho‐leitosos, vasos polimórficos e vasos lineares irregulares,20 refletindo o rápido crescimento do tumor.
A coloração com hematoxilina & eosina revela a proliferação de pequenas células tumorais basofílicas na derme e/ou hipoderme,21 com citoplasma escasso, mitoses abundantes e grânulos citoplasmáticos densos. Células necróticas também são comuns.8,20 Exemplos da histopatologia estão representados na figura 3.
 Cordões de células tumorais na derme (aumento de 200×). (B) As células tumorais apresentam citoplasma escasso e núcleos redondos ou irregulares (aumento de 400×).")
A citoqueratina 20 (CK20) e marcadores neuroendócrinos, como cromogranina A, sinaptofisina, CD56, enolase neurônio‐específica e neurofilamento são expressos no CCM.3,14,21 A imunopositividade paranuclear semelhante a pontos para CK20 é altamente sugestiva, enquanto os marcadores neuroendócrinos não são específicos.3
Para pacientes com linfonodos clinicamente negativos, a BLS deve ser realizada e sua taxa de positividade varia de 30% a 38%.14 Quando negativo, menor risco de recorrência,22 melhor sobrevida livre de doença (SLD) e sobrevida global (SG) são observados.14
O uso de exames de imagens para investigação diagnóstica de pacientes com CCM ainda está em discussão. O NCCN não recomenda exames de imagem em pacientes sem suspeita clínica de linfonodos acometidos. A BLS é considerada o teste mais confiável para identificar metástases em linfonodos não clinicamente suspeitos. A ressonância magnética cerebral, as tomografias computadorizadas (TC) de pescoço, tórax, abdome e pelve ou PET‐CT são indicadas para planejamento cirúrgico ou quando houver suspeita de tumor irressecável ou metástases a distância.14
Gupta et al. mostraram que a TC tem baixa sensibilidade (em torno de 20%) para detecção de metástases linfonodais e baixa especificidade para metástases a distância.22 Portanto, embora a TC tenha sido usada como ferramenta de triagem para metástases regionais ou a distância em CCM, os dados que suportam sua aplicação são limitados.14,22
O PET‐CT tem sido amplamente estudado em pacientes com CCM.14 Colgan et al. avaliaram retrospectivamente 36 pacientes com CCM que realizaram exames de PET‐CT antes da BLS e observaram 83% e 95% de sensibilidade e especificidade, respectivamente.23
Diagnóstico diferencialClinicamente, os diagnósticos diferenciais para CCM são lesões benignas da pele, como cisto acneiforme, lipoma, dermatofibroma ou fibroma e lesão vascular. Lesões malignas representadas por câncer de pele não melanoma, linfoma cutâneo, carcinoma metastático e sarcoma também devem ser consideradas diagnósticos diferenciais.15
Histologicamente, o diagnóstico também pode ser um desafio, pois o CCM é semelhante a uma variedade de outros tumores que têm células azuis pequenas e arredondadas, incluindo linfomas, melanoma amelanótico, metástases cutâneas de carcinoma de células pequenas de pulmão3 e outros carcinomas metastáticos (neuroblastoma, rabdomiossarcoma, tumor desmoplásico de pequenas células, condrossarcoma mesenquimal, sarcoma de Ewing e osteossarcoma).8 O CCM in situ, no qual as células neoplásicas são limitadas à epiderme e/ou epitélio folicular, pode ser confundido com carcinoma de células escamosas in situ, melanoma in situ ou outra neoplasia intraepidérmica pagetoide.20
Em decorrência dos aspectos morfológicos similares, o diagnóstico diferencial mais desafiador é o câncer de pulmão de pequenas células metastático3,8 que exige painel de marcadores imuno‐histoquímicos para definir o diagnóstico. CK20 e o fator de transcrição da tireoide 1 (TTF‐1) têm maior sensibilidade e especificidade para excluir câncer de pulmão de pequenas células;8 CK20 é positivo em 70% a 100% do CCM e negativo em câncer de pulmão de pequenas células, enquanto TTF‐1 é sempre negativo em CCM e positivo em mais de 80% dos cânceres de pulmão de pequenas células.3,8,14
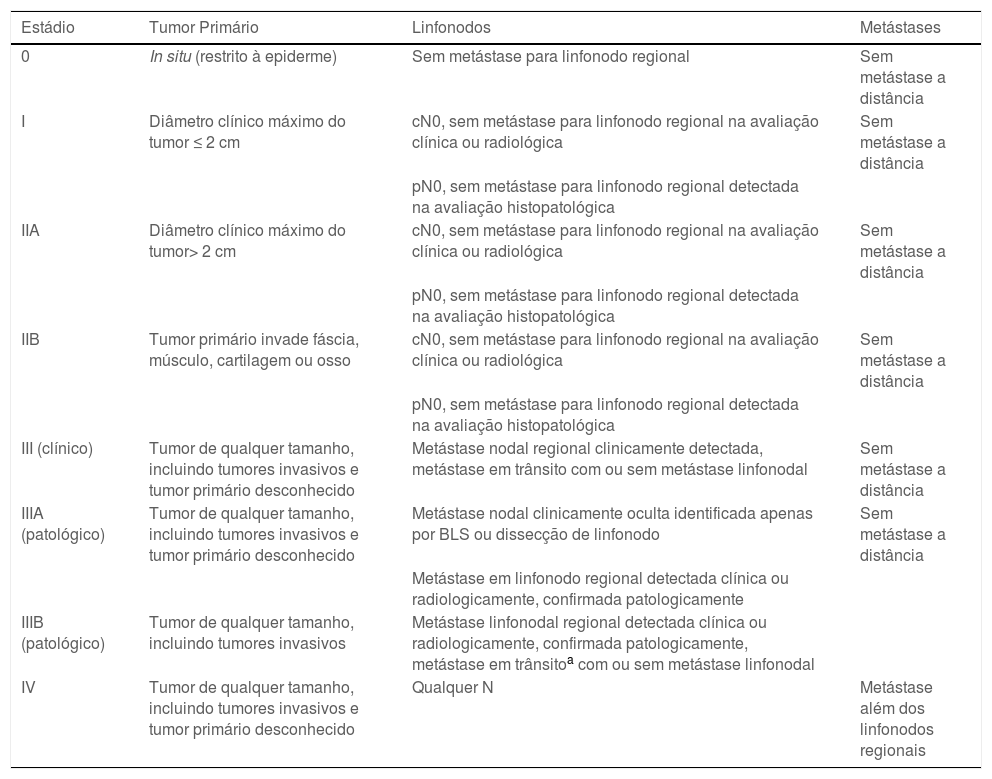
EstadiamentoA atual 8ª edição do sistema de estadiamento do American Joint Committee on Cancer (AJCC) é baseada em uma análise atualizada de 9.387 casos de CCM do National Cancer Database, com acompanhamento médio de 28,2 meses (tabela 1).24 De acordo com o AJCC, os seguintes parâmetros são necessários para o estadiamento do CCM T: 1) diâmetro máximo do tumor medido clinicamente, antes da ressecção e 2) extensão do tumor (fáscia, músculo, cartilagem ou invasão óssea).24 O diâmetro do tumor tem valor prognóstico, estando significantemente associado ao envolvimento de linfonodos, sobrevida doença‐específica (SDE) e SG.14
Estadiamento do carcinoma de células de Merkel – AJCC 8ª edição
| Estádio | Tumor Primário | Linfonodos | Metástases |
|---|---|---|---|
| 0 | In situ (restrito à epiderme) | Sem metástase para linfonodo regional | Sem metástase a distância |
| I | Diâmetro clínico máximo do tumor ≤ 2 cm | cN0, sem metástase para linfonodo regional na avaliação clínica ou radiológica | Sem metástase a distância |
| pN0, sem metástase para linfonodo regional detectada na avaliação histopatológica | |||
| IIA | Diâmetro clínico máximo do tumor> 2 cm | cN0, sem metástase para linfonodo regional na avaliação clínica ou radiológica | Sem metástase a distância |
| pN0, sem metástase para linfonodo regional detectada na avaliação histopatológica | |||
| IIB | Tumor primário invade fáscia, músculo, cartilagem ou osso | cN0, sem metástase para linfonodo regional na avaliação clínica ou radiológica | Sem metástase a distância |
| pN0, sem metástase para linfonodo regional detectada na avaliação histopatológica | |||
| III (clínico) | Tumor de qualquer tamanho, incluindo tumores invasivos e tumor primário desconhecido | Metástase nodal regional clinicamente detectada, metástase em trânsito com ou sem metástase linfonodal | Sem metástase a distância |
| IIIA (patológico) | Tumor de qualquer tamanho, incluindo tumores invasivos e tumor primário desconhecido | Metástase nodal clinicamente oculta identificada apenas por BLS ou dissecção de linfonodo | Sem metástase a distância |
| Metástase em linfonodo regional detectada clínica ou radiologicamente, confirmada patologicamente | |||
| IIIB (patológico) | Tumor de qualquer tamanho, incluindo tumores invasivos | Metástase linfonodal regional detectada clínica ou radiologicamente, confirmada patologicamente, metástase em trânsitoa com ou sem metástase linfonodal | |
| IV | Tumor de qualquer tamanho, incluindo tumores invasivos e tumor primário desconhecido | Qualquer N | Metástase além dos linfonodos regionais |
Fonte adaptada: Harms KL et al., 2016.24.
O CCM é caracteristicamente um tumor agressivo e localmente invasivo, com alta incidência de recorrência local e envolvimento de linfonodos regionais.1 A BLS é importante ferramenta de estadiamento recomendada para todos os pacientes com linfonodo clinicamente negativo, sempre que possível, para realizar o estadiamento nodal patológico.14
De acordo com a 8ª edição do sistema de estadiamento AJCC, as estimativas de SG de cinco anos para o estadiamento clínico e patológico da doença local foram 45,0% e 62,8% para o estádio I; 30,9% e 54,6% para o estádio IIA e 27,3% e 34,8% para o estádio IIB, respectivamente. A SG de cinco anos para o estádio IIIA revisado foi de 40,3%, enquanto para o estádio IIIB foi de 26,8%.24 Na doença em estádio IV, a taxa de sobrevida de dois anos é de apenas 26%.1
Sihto et al., em 2009, relataram que os CCM positivos para o poliomavírus apresentaram maior taxa de sobrevida em comparação com os tumores negativos para esse vírus; a sobrevida em cinco anos foi de 45% vs. 13% (p‐valor<0,01), respectivamente.12
TratamentoCirurgia para o tumor primárioA cirurgia é a modalidade de tratamento inicial para a maioria dos casos de CCM. A definição das margens cirúrgicas ainda é um tema controverso.14 O objetivo do tratamento cirúrgico é uma margem histológica negativa e, em geral, a excisão cirúrgica com margens de 1 cm a 2 cm continua a ser o primeiro passo no manejo do tumor.25 Boyer et al. analisaram 45 pacientes com CCM em estádio I e relataram margem mediana de 16,7 mm de pele clinicamente normal necessária para a ausência de envolvimento histológico.26
Um estudo de 1.795 pacientes com CCM estádios I e II não mostrou diferenças entre a excisão cirúrgica ampla e a cirurgia micrográfica de Mohs em relação à presença de tumor residual nas margens cirúrgicas, e nenhuma diferença na SG entre as duas modalidades de tratamento foi encontrada.27 As margens histologicamente negativas estão relacionadas ao melhor controle local e sobrevida, principalmente nos casos de doença localizada tratada apenas com cirurgia. Em todos os casos de tratamento cirúrgico, a BLS deve ser planejada antes da excisão definitiva, pois a cirurgia pode alterar a drenagem linfática.14
Santamaria‐Barria et al. elucidaram o papel da BLS no manejo do CCM em uma revisão de 161 pacientes, tendo identificado micrometástases em 1/3 deles.28 Assim, após verificação de margens livres e, se indicado BLS, o desafio é determinar se deve ou não oferecer tratamento adjuvante.
RadioterapiaA radioterapia (RT) tem papel importante no tratamento do CCM. O CCM é uma neoplasia radiossensível, e a RT pode ser considerada terapia primária em pacientes que não são candidatos à cirurgia.29,30 Embora a RT por si só seja inferior à ressecção cirúrgica em razão do risco de recorrência de CCM a distância, ela pode ser usada quando a cirurgia é contraindicada31–33 com bom controle locorregional e SG em cinco anos de até 40% a 60% em pacientes com metástases macroscópicas primárias e/ou nodais.33
Em relação a seu uso como terapia adjuvante, os dados são conflitantes. A RT adjuvante pós‐operatória parece aumentar o controle local do tumor, mas não tem impacto significante na sobrevida global relacionada ao tumor.34,35 Em uma revisão com casos de CCM apenas de cabeça e pescoço, a RT adjuvante apresentou benefício significante na sobrevida em relação à cirurgia sozinha, e a quimioterapia (QT) associada à RT ofereceu vantagem sobre a RT sozinha em casos de tumores grandes (> 3 cm) ou margens positivas.36 Uma análise retrospectiva de CCM estádios I a III mostrou baixa taxa de recorrência (3%) em pacientes com linfonodo clinicamente negativo tratados com cirurgia adequada (incluindo BLS) e o uso seletivo de RT adjuvante para tumores de alto risco, incluindo invasão linfovascular; segunda malignidade (leucemia/linfoma) no momento do diagnóstico, múltiplos linfonodos positivos e extensão extracapsular; tumores >2 cm. Os autores também concluem que, com cirurgia adequada, é improvável que o uso rotineiro de RT local adjuvante seja benéfico para a grande maioria dos pacientes.37 Além disso, alguns dados sugeriram que a RT adjuvante pode ser omitida para aqueles com pequenos tumores primários (< 1 cm), margens cirúrgicas livres, BLS negativa e ausência de fatores de mau prognóstico, como invasão linfovascular e imunodeficiência.38
Uma análise retrospectiva de 46 casos de CCM de cabeça e pescoço de baixo risco tratados entre 2006 e 2015 demonstrou que a não realização da RT pós‐operatória foi associada a risco significantemente maior de recorrência local (recorrência local em cinco anos de 26% no grupo tratado sem RT adjuvante versus 0% no grupo que recebeu RT adjuvante; p=0,02). Os pacientes foram considerados de baixo risco em caso de tumor primário ≤ 2,0 cm de diâmetro, margens microscopicamente negativas na excisão cirúrgica, BLS negativa e ausência de imunossupressão crônica.39
Jouary Y et al. mostraram redução significante na recorrência local sem melhora da sobrevida global com RT adjuvante em estudo realizado antes da introdução da BLS no tratamento do CCM.40
Em uma coorte retrospectiva e não randomizada de 6.908 casos do National Cancer Database dos EUA, a RT adjuvante foi associada a melhora da sobrevida em pacientes com CCM localizado (estádios I e II) em comparação com pacientes que passaram apenas pelo tratamento cirúrgico (estádio I: hazard ratio [HR=0,71]; 95%IC 0,64%‐0,80%; p <0,001; estádio II: HR=0,77; 95%IC 0,66%‐0,89%; p <0,001). No entanto, em pacientes com envolvimento nodal (estádio III), não houve diferença estatisticamente significante na SG entre os pacientes que tiveram cirurgia seguida de RT em comparação com a cirurgia isolada (HR=0,98; 95%IC 0,86%‐1,12; p=0,80).41
Em contraste, um estudo retrospectivo do Moffitt Cancer Center, avaliando 171 pacientes, encontrou melhora no controle locorregional e SG entre pacientes com linfonodos patologicamente ou clinicamente positivos tratados com RT pós‐operatória. Nessa publicação, os pacientes foram tratados com excisão local ampla (margens de 1‐2 cm) e todos realizaram estadiamento nodal.42
Assim, o NCCN incluiu a RT como opção de tratamento adjuvante para todos os estádios do CCM e, idealmente, deve ser realizada em quatro a seis semanas após a cirurgia, pois o atraso tem sido associado a piores desfechos.14
Tratamento sistêmicoQuimioterapiaO papel da QT adjuvante para o CCM é menos definido. Hasan et al. realizaram uma grande revisão sistemática e notaram que a RT adjuvante resultou em taxas de controle local em três anos significantemente maiores, taxas de recorrência diminuídas e taxas de SG melhores em três anos, enquanto a QT adjuvante não ofereceu nenhum benefício.43 Além disso, estudos adicionais sugerem que o efeito da QT adjuvante na recorrência não é claro ou que não há melhora significante na sobrevida quando comparada à RT adjuvante isolada.29,44,45
Por muitos anos, a QT foi a única opção disponível como tratamento para a doença avançada. O CCM é considerado tumor quimiossensível, e esquemas de platina com etoposídeo ou taxanos e antraciclinas isoladas ou em combinação são inicialmente eficazes, com taxa de resposta de até 75%, mas geralmente a duração da resposta é curta, não resultando em benefício de sobrevida. A sobrevida livre de progressão (SLP) mediana varia de três a oito meses, e a duração da resposta parcial é de apenas três meses para a terapia de primeira linha.25,46 Dada a idade avançada de muitos pacientes e suas comorbidades, a toxicidade da QT pode afetar gravemente a qualidade de vida.
ImunoterapiaAs imunoterapias estão atualmente em foco, uma vez que foi descrita a importância do sistema imunológico no controle do CCM; os inibidores de PD‐1 (programmed cell death 1) e PD‐L1 (programmed cell death protein ligand 1) são opções promissoras nos casos de doença metastática.21
As vias PD‐1 e PD‐L1/PD‐L2 formam um sistema complexo de receptores e ligantes envolvidos no controle da ativação de células T. Na maioria dos tumores, PD‐L1 é predominantemente expresso em células tumorais e PD‐1 nos linfócitos que se infiltram no tumor.1 No entanto, o PD‐1 pode ser expresso não apenas por linfócitos T CD8+, mas também pelas células CD4+, CD20+, Treg e células NK.47 A ligação entre PD‐1 e seu ligante PD‐L1 leva à inativação e diminuição da proliferação de células T. Esse processo parece ser um mecanismo importante para a inibição da resposta imune pelo tumor.48
A associação da imunossupressão com o maior risco de CCM, além de alguns dados que mostram melhor prognóstico em tumores com alta infiltração de linfócitos T CD8+, tornou‐se uma justificativa para o uso da imunoterapia.15,49 Além disso, Lipson et al., em 2013, relataram associação significante entre a presença do MCPyV e a expressão de PD‐L1 no tumor e entre a presença do MCPyV com um infiltrado inflamatório moderado a intenso. Esses achados sugerem a hipótese de que uma resposta imune a antígenos virais cria um ambiente pró‐inflamatório local que estimula a expressão de PD‐L1 no tumor.50 Essas descobertas abriram caminho para o uso de inibidores de checkpoint imunes no MCC metastático.
O primeiro estudo nesse cenário foi o JAVELIN Merkel 200, um ensaio clínico multicêntrico de fase II, aberto, que investigou a atividade clínica e a segurança de avelumabe, um anticorpo anti‐PD‐L1. O estudo apresenta parte A, realizado em pacientes com CCM previamente expostos à QT,51 e parte B, realizado em pacientes que não haviam recebido tratamento sistêmico prévio para doença metastática.52 Na parte A, 88 pacientes foram tratados com avelumabe e a taxa de resposta global confirmada (ORR) foi de 33% (incluindo 10 respostas completas) e com 26% dos pacientes sem progressão em dois anos. A duração mediana da resposta não foi alcançada, e a SG mediana foi de 12,6 meses.51 Uma resposta objetiva de 34,5% foi encontrada em PD‐L1 positivo contra apenas 18,8% em pacientes PDL‐1 negativos (expressão <1%.) Considerando o corte na expressão de PD‐L1 em 5%, 52,6% dos pacientes PD‐L1 positivos versus 23,6% dos pacientes PD‐L1 negativos tiveram resposta objetiva. Além disso, 26,1% dos pacientes com tumores MCPyV‐positivos em comparação com 35,5% nos tumores MCPyV‐negativos tiveram resposta.53 Outra análise de subgrupo sugeriu maior probabilidade de resposta em pacientes que receberam menos terapias sistêmicas prévias, o que foi confirmado na parte B do ensaio Javelin Merkel 200. A resposta objetiva entre os 29 pacientes incluídos no cenário de primeira linha foi de 62%, com proporção de respostas com duração de seis meses ou mais de 83%.52
Munhoz et al., em 2020, descreveram a experiência com avelumabe como tratamento de segunda linha (ou primeira linha em pacientes não candidatos ao uso de QT) em um subconjunto de 46 pacientes latino‐americanos (idade média 71,6 anos; 60,9% homens; duração média do tratamento 7,9 meses). Respostas objetivas foram observadas em 19 pacientes (resposta objetiva 57,9%; resposta completa 15,8%; resposta parcial 42,1%; e doença estável 10,5%) e segurança consistente com dados globais.54
Outro estudo com características semelhantes demonstrou a eficácia do pembrolizumabe no CCM, um anticorpo monoclonal anti‐PD‐1. Um total de 50 pacientes com CCM avançado foram tratados com resposta objetiva de 56% e taxa de SLP em seis meses de 67%. Em contraste com o estudo com avelumabe, uma taxa de resposta mais alta foi observada nos tumores positivos para MCPyV (62%) do que nos tumores negativos para MCPyV (44%).55
No estudo CheckMate 358, o nivolumabe, outro anti‐PD‐1, foi avaliado em 25 pacientes com CCM avançado, uma resposta objetiva de 64%. As respostas ocorreram em pacientes virgens de tratamento (71%), bem como naqueles com pelo menos uma terapia sistêmica anterior (63%), com tempo médio curto para resposta (75% dos pacientes responderam em dois meses).56
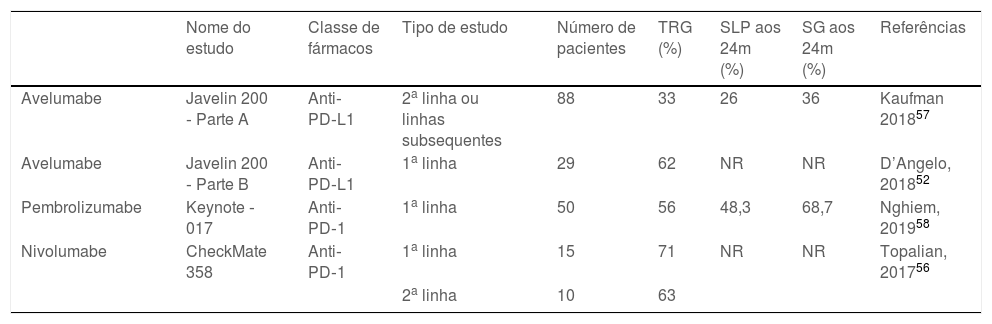
O resumo dos recentes estudos com imunoterapia para o tratamento do MCC metastático pode ser visto na tabela 2.52,56–58
Dados importantes de ensaios clínicos recentes envolvendo imunoterapia52,56–58
| Nome do estudo | Classe de fármacos | Tipo de estudo | Número de pacientes | TRG (%) | SLP aos 24m (%) | SG aos 24m (%) | Referências | |
|---|---|---|---|---|---|---|---|---|
| Avelumabe | Javelin 200 ‐ Parte A | Anti‐PD‐L1 | 2a linha ou linhas subsequentes | 88 | 33 | 26 | 36 | Kaufman 201857 |
| Avelumabe | Javelin 200 ‐ Parte B | Anti‐PD‐L1 | 1a linha | 29 | 62 | NR | NR | D’Angelo, 201852 |
| Pembrolizumabe | Keynote ‐ 017 | Anti‐PD‐1 | 1a linha | 50 | 56 | 48,3 | 68,7 | Nghiem, 201958 |
| Nivolumabe | CheckMate 358 | Anti‐PD‐1 | 1a linha | 15 | 71 | NR | NR | Topalian, 201756 |
| 2a linha | 10 | 63 |
TRG, taxa de resposta global; SLP, sobrevida livre de progressão; SG, sobrevida global; NR, não relatado.
Dada a eficácia da imunoterapia para CCM metastático, o próximo desafio consiste em avaliar tais tratamentos no cenário adjuvante. Em razão do alto risco de recorrência do CCM, apesar do tratamento inicial e da falta de benefício do tratamento citotóxico, sua investigação é altamente relevante, cobrindo uma necessidade médica não atendida. Dois estudos de fase II com nivolumabe e avelumabe estão avaliando o uso de inibidores de checkpoints imunes para pacientes de alto risco, ainda sem os resultados oficialmente publicados. O cenário neoadjuvante com o estudo CheckMate 358, envolvendo 39 pacientes com CCM ressecável estágio IIA a IV, avaliou o uso do nivolumabe (≥ 1 dose) cerca de quatro semanas antes da cirurgia. Três pacientes não foram operados por progressão tumoral e 36 pacientes foram operados, dentre os quais 17 (42,2%) obtiveram resposta patológica completa. Entre 33 pacientes avaliáveis radiograficamente submetidos à cirurgia, 18 (54,5%) tiveram redução tumoral ≥ 30%. Nenhum paciente com resposta patológica completa teve recidiva tumoral durante a observação.59
ConclusãoO CCM é doença rara e agressiva, cuja incidência está aumentando. Nas últimas décadas, muitos avanços foram feitos no conhecimento da biologia, diagnóstico e tratamento do CCM, mas grandes desafios ainda permanecem. Os tumores primários devem ser tratados com excisão cirúrgica completa, com margens cirúrgicas adequadas. RT adjuvante deve ser considerada. Em relação ao CCM avançado, a imunoterapia mudou drasticamente o padrão de tratamento, uma vez que é superior à QT. Notáveis respostas foram observadas em pacientes com CCM tratados com inibidores do eixo PD‐1/PD‐L1 e parece ser maneira promissora de mudar o prognóstico desse câncer de pele grave.
Suporte financeiroNenhum.
Contribuição dos autoresStella Meireles Siqueira: Análise estatística; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Gabriella Campos‐do‐Carmo: Análise estatística; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Alexssandra Lima Siqueira dos Santos: Análise estatística; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Cícero Martins: Análise estatística; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Andreia Cristina de Melo: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Siqueira SM, Campos‐do‐Carmo G, Santos ALS, Martins C, Melo AC. Merkel cell carcinoma: epidemiology, clinical features, diagnosis and treatment of a rare disease. An Bras Dermatol. 2023;98:277–86.
Trabalho realizado no Instituto Nacional de Câncer, Rio de Janeiro, RJ, Brasil.
