


Os linfomas cutâneos de células T constituem um grupo heterogêneo de doenças linfoproliferativas, caracterizadas por infiltração da pele por células T maduras malignas. A micose fungoide é a forma mais comum de linfoma cutâneo de células T, representando mais de 60% dos casos. A micose fungoide no estágio inicial é geralmente indolente, progredindo lentamente de algumas manchas ou placas para envolvimento cutâneo mais generalizado. Entretanto, 20% a 25% dos pacientes progridem para estágios avançados, com desenvolvimento de tumores cutâneos, disseminação extracutânea e mau prognóstico. As modalidades de tratamento podem ser divididas em dois grupos: terapias direcionadas à pele e terapias sistêmicas. As terapias direcionadas à pele incluem agentes tópicos, fototerapia e radioterapia. As terapias sistêmicas incluem modificadores da resposta biológica, imunoterapias e agentes quimioterápicos. Para a micose fungoide em estágio inicial, as terapias direcionadas à pele são preferidas, para controlar a doença, melhorar os sintomas e a qualidade de vida. Na micose fungoide refratária em estágio inicial e na doença em estágio avançado, é necessário tratamento sistêmico. Neste artigo, apresentamos uma compilação das opções atuais de tratamento para a micose fungoide e síndrome de Sézary.
Os linfomas cutâneos primários são um grupo heterogêneo de linfomas não‐Hodgkin que se apresentam na pele sem evidência de doença extracutânea no momento do diagnóstico. Representam 19% dos pacientes com linfomas extranodais, com incidência anual nos países ocidentais de 1 caso por 100.000 mil habitantes‐ano. Os linfomas cutâneos primários são classificados pela Organização Mundial da Saúde – Organização Europeia para Pesquisa e Tratamento do Câncer (OMS‐EORTC) em linfomas cutâneos de células T (LCCT) e linfomas cutâneos de células B (LCCB). No ocidente, os LCCT representam aproximadamente 75% a 80% de todos os linfomas cutâneos primários.1,2
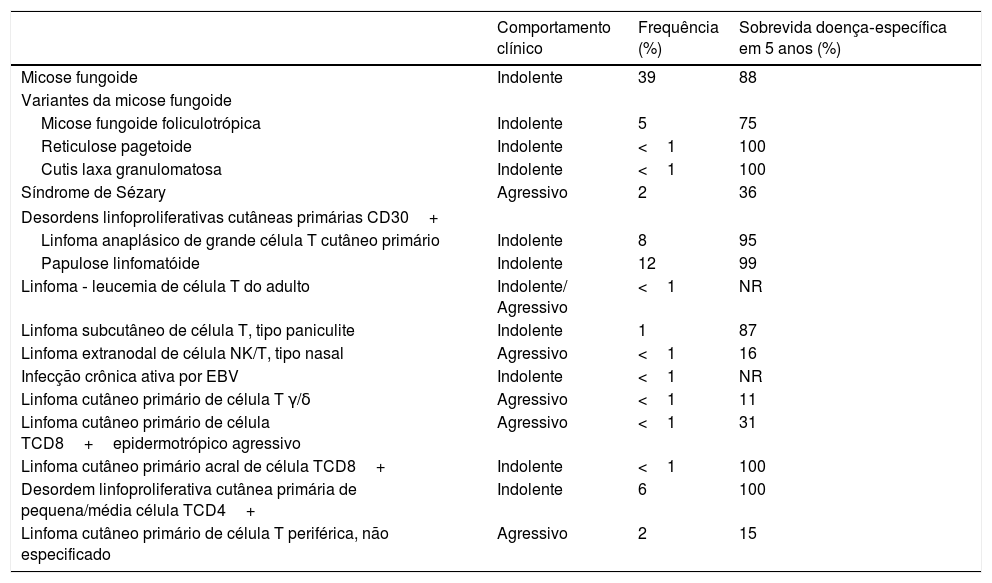
Os LCCT aparecem primariamente na pele e podem progredir para os linfonodos, sangue e vísceras. Constituem um grupo heterogêneo, com considerável variação na apresentação clínica, bem como nas características histológicas, imunofenotípicas e prognósticas. A classificação atualizada está mostrada na tabela 1.1
Classificação dos linfomas de células T com manifestações cutâneas primárias de acordo com seu comportamento clínico, frequência e sobrevivência doença‐específica em 5 anos. Baseada nas diretrizes OMS‐EORTC1
| Comportamento clínico | Frequência (%) | Sobrevida doença‐específica em 5 anos (%) | |
|---|---|---|---|
| Micose fungoide | Indolente | 39 | 88 |
| Variantes da micose fungoide | |||
| Micose fungoide foliculotrópica | Indolente | 5 | 75 |
| Reticulose pagetoide | Indolente | <1 | 100 |
| Cutis laxa granulomatosa | Indolente | <1 | 100 |
| Síndrome de Sézary | Agressivo | 2 | 36 |
| Desordens linfoproliferativas cutâneas primárias CD30+ | |||
| Linfoma anaplásico de grande célula T cutâneo primário | Indolente | 8 | 95 |
| Papulose linfomatóide | Indolente | 12 | 99 |
| Linfoma ‐ leucemia de célula T do adulto | Indolente/ Agressivo | <1 | NR |
| Linfoma subcutâneo de célula T, tipo paniculite | Indolente | 1 | 87 |
| Linfoma extranodal de célula NK/T, tipo nasal | Agressivo | <1 | 16 |
| Infecção crônica ativa por EBV | Indolente | <1 | NR |
| Linfoma cutâneo primário de célula T γ/δ | Agressivo | <1 | 11 |
| Linfoma cutâneo primário de célula TCD8+epidermotrópico agressivo | Agressivo | <1 | 31 |
| Linfoma cutâneo primário acral de célula TCD8+ | Indolente | <1 | 100 |
| Desordem linfoproliferativa cutânea primária de pequena/média célula TCD4+ | Indolente | 6 | 100 |
| Linfoma cutâneo primário de célula T periférica, não especificado | Agressivo | 2 | 15 |
NR, não relatado.
Os LCCT também podem ser classificados de acordo com a sua agressividade, conforme descrito na tabela 1. Os linfomas indolentes, habitualmente, apresentam um curso crônico com recidivas frequentes. A doença é geralmente considerada incurável apesar do tratamento, com sobrevida média de 5 a 10 anos. No entanto, alguns pacientes podem sobreviver mais de 20 anos. Os linfomas agressivos apresentam progressões mais rápidas quando comparados às formas indolentes e podem levar à morte em questão de meses.1
Este artigo aborda as possibilidades terapêuticas para a micose fungoide (MF) e a síndrome de Sézary (SS). A MF é o tipo mais comum e responde por 60% dos LCCT e quase 50% de todos os linfomas cutâneos primários. A maioria dos pacientes é de adultos ou idosos, com proporção de homens para mulheres de 2:1 e incidência mundial de cerca de 5‐6 casos por milhão de habitantes‐ano. A SS é uma variante leucêmica rara de LCCT.1,2
Micose fungoide e síndrome de SézaryA MF é um linfoma não‐Hodgkin de células T maduras com apresentação primária na pele, mas com potencial envolvimento secundário dos linfonodos, sangue e vísceras. As lesões cutâneas incluem manchas (patches) ou placas que podem estar localizadas ou disseminadas, tumores e eritrodermia. O curso da MF é variável. Alguns pacientes mantêm a doença limitada na pele por muitos anos; outros podem evoluir mais rapidamente para o envolvimento extracutâneo com pior prognóstico. A SS é uma forma eritrodérmica e pruriginosa de LCCT, caracterizada por linfadenopatia periférica e pela presença de células T neoplásicas com núcleos cerebriformes (células de Sézary), clonalmente relacionadas, na pele, linfonodos e sangue periférico.2
Na histologia, é tipicamente um linfoma epidermotrópico de linfócitos de tamanho pequeno a médio com núcleos cerebriformes. As células neoplásicas têm um fenótipo de células T maduras, CD3+, CD4+, CD45RO+, CD8−, com perda variável da expressão de CD7.1 A molécula CD30 pode eventualmente ser expressa nas células neoplásicas, mas essa expressão parece ser mais frequente e mais intensa nos casos que mostram transformação para linfoma de células grandes. A MF transformada é definida pela presença de células grandes (≥ 4 vezes o tamanho de um pequeno linfócito) em quantidade superior a 25% do infiltrado dérmico ou pela formação de nódulos microscópicos, e geralmente está associada a um pior prognóstico.3
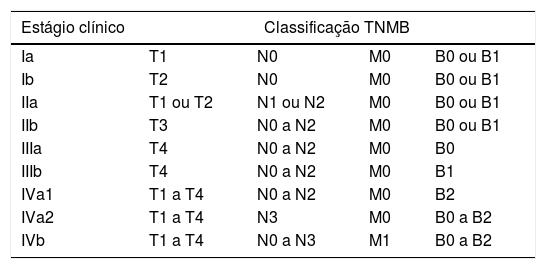
O estadiamento clínico da MF e SS é baseado na classificação do acometimento cutâneo (T), linfonodal (N), visceral (M) e hematológico (B) (tabelas 2 e 3). A classificação do TNMB deve incluir o exame cuidadoso da pele (com atenção ao couro cabeludo, palmas, plantas e períneo), biópsia(s) de lesão(ões) de pele, hemograma completo com pesquisa de células de Sézary, citometria de fluxo do sangue periférico, exames bioquímicos e estudos de imagem.2 Como o Brasil é endêmico para o HTLV‐1, a triagem para esse retrovírus é obrigatória para todos os pacientes com suspeita ou comprovação de LCCT, para diferenciar MF/SS de linfoma‐leucemia de células‐T do adulto.4
Classificação revisada do TNMB para micose fungoide e síndrome de Sézary2,5
| Pele (T) | |
| T1 | Manchas (patches)/placas limitadas (envolvendo <10% da superfície total da pele) |
| T1a | Apenas patches |
| T1b | Placas±patches |
| T2 | Manchas (patches)/placas generalizadas (envolvendo ≥ 10% da superfície total da pele) |
| T2a | Apenas patches |
| T2b | Placas±patches |
| T3 | Tumor (es) |
| T4 | Eritrodermia |
| Linfonodo (N) | |
| N0 | Sem linfonodos periféricos anormais do ponto de vista clínico |
| N1 | Linfonodos periféricos clinicamente anormais; linfadenite dermatopática ou envolvimento histológico por linfócitos atípicos isolados sem alteração da arquitetura linfonodal |
| N1a | Clone negativo |
| N1b | Clone positivo |
| N2 | Linfonodos periféricos clinicamente anormais; envolvimento histológico por agregados de linfócitos atípicos sem alteração da arquitetura linfonodal |
| N2a | Clone negativo |
| N2b | Clone positivo |
| N3 | Linfonodos periféricos clinicamente anormais; comprometimento histológico franco com alteração parcial ou total da arquitetura linfonodal |
| NX | Linfonodos periféricos clinicamente anormais; sem confirmação histológica |
| Vísceras (M) | |
| M0 | Sem envolvimento visceral |
| M1 | Com envolvimento visceral |
| Sangue (B) | |
| B0 | Não há células atípicas (Sézary) circulantes (ou <5% de linfócitos atípicos) |
| B0a | Clone negativo |
| B0b | Clone positivo |
| B1 | Baixa carga tumoral no sangue (≥ 5% dos linfócitos são células Sézary, mas não B2) |
| B1a | Clone negativo |
| B1b | Clone positivo |
| B2 | Elevada carga tumoral no sangue e rearranjo clonal do TCR (≥ 1.000 células de Sézary/microL. e/ou CD4:CD8 ≥ 10, e/ou CD4+CD7 ‐ ≥ 40%, e/ou CD4+CD26 ‐ ≥30%) |
Sistema de estadiamento clínico para micose fungoide e síndrome de Sézary2
| Estágio clínico | Classificação TNMB | |||
|---|---|---|---|---|
| Ia | T1 | N0 | M0 | B0 ou B1 |
| Ib | T2 | N0 | M0 | B0 ou B1 |
| IIa | T1 ou T2 | N1 ou N2 | M0 | B0 ou B1 |
| IIb | T3 | N0 a N2 | M0 | B0 ou B1 |
| IIIa | T4 | N0 a N2 | M0 | B0 |
| IIIb | T4 | N0 a N2 | M0 | B1 |
| IVa1 | T1 a T4 | N0 a N2 | M0 | B2 |
| IVa2 | T1 a T4 | N3 | M0 | B0 a B2 |
| IVb | T1 a T4 | N0 a N3 | M1 | B0 a B2 |
T, pele; N, linfonodo; M, víscera; B, sangue periférico.
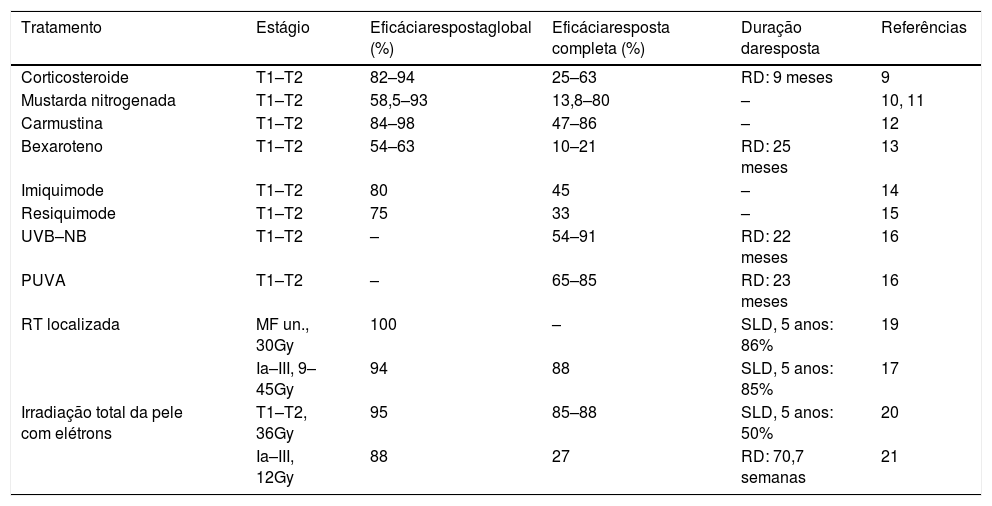
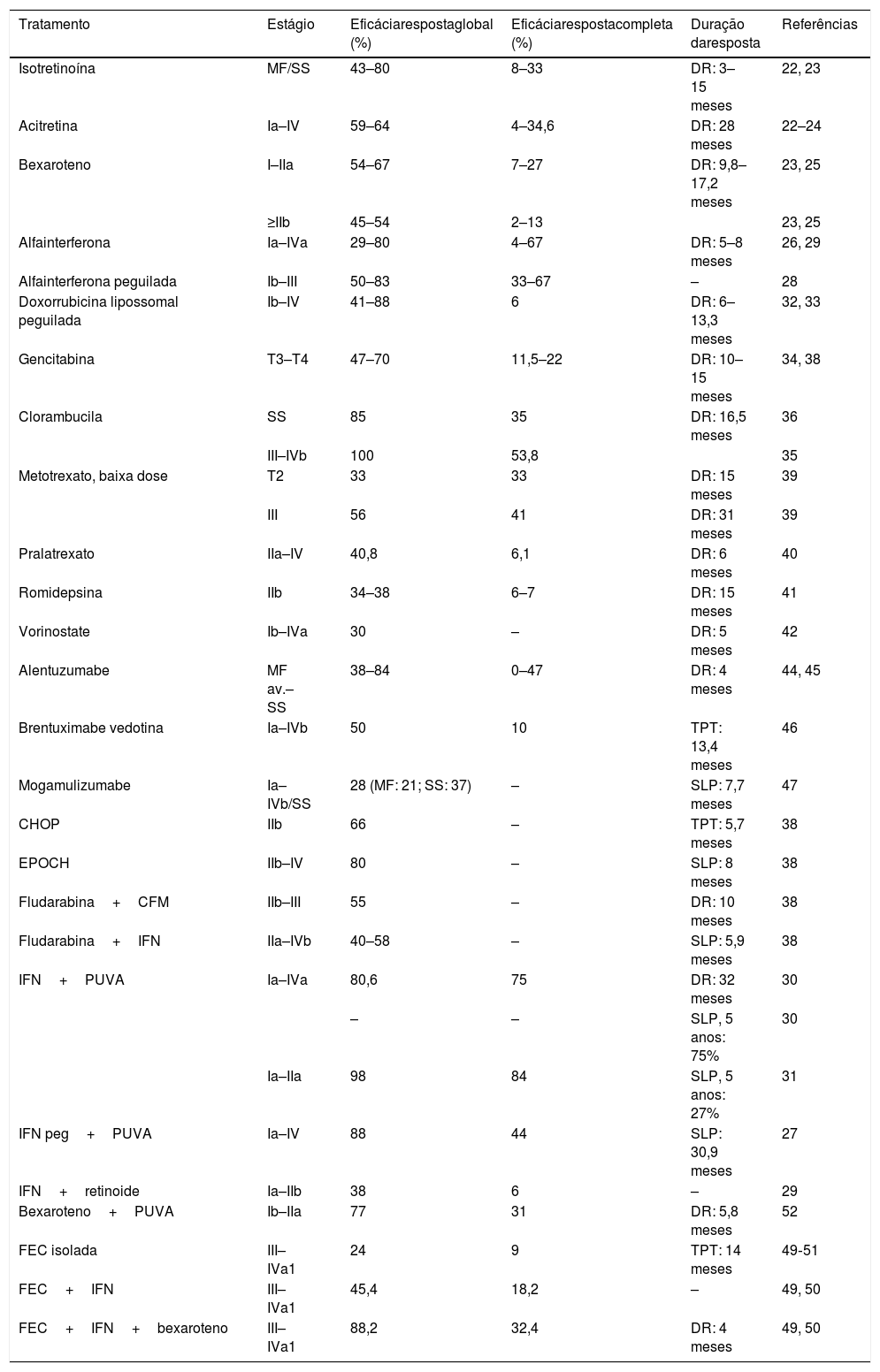
A maioria dos tratamentos disponíveis para MF/SS raramente induz remissões a longo prazo. Os resultados dos tratamentos que devem ser considerados incluem taxa de resposta global (taxa de remissão completa+taxa de remissão parcial), remissão completa, remissão parcial (remissão de pelo menos 50% da carga de doença), duração da resposta, tempo até o próximo tratamento, sobrevida livre de progressão, sobrevida global, sobrevida doença‐específica e melhora dos sintomas e da qualidade de vida (tabelas 4 e 5). Tanto o tratamento quanto o prognóstico variam de acordo com os estágios da doença.6
Taxas de resposta e duração das respostas para os tratamentos dirigidos à pele para a MF/SS
| Tratamento | Estágio | Eficáciarespostaglobal (%) | Eficáciaresposta completa (%) | Duração daresposta | Referências |
|---|---|---|---|---|---|
| Corticosteroide | T1–T2 | 82–94 | 25–63 | RD: 9 meses | 9 |
| Mustarda nitrogenada | T1–T2 | 58,5–93 | 13,8–80 | – | 10, 11 |
| Carmustina | T1–T2 | 84–98 | 47–86 | – | 12 |
| Bexaroteno | T1–T2 | 54–63 | 10–21 | RD: 25 meses | 13 |
| Imiquimode | T1–T2 | 80 | 45 | – | 14 |
| Resiquimode | T1–T2 | 75 | 33 | – | 15 |
| UVB–NB | T1–T2 | – | 54–91 | RD: 22 meses | 16 |
| PUVA | T1–T2 | – | 65–85 | RD: 23 meses | 16 |
| RT localizada | MF un., 30Gy | 100 | – | SLD, 5 anos: 86% | 19 |
| Ia–III, 9–45Gy | 94 | 88 | SLD, 5 anos: 85% | 17 | |
| Irradiação total da pele com elétrons | T1–T2, 36Gy | 95 | 85–88 | SLD, 5 anos: 50% | 20 |
| Ia–III, 12Gy | 88 | 27 | RD: 70,7 semanas | 21 |
SLP, sobrevida livre de progressão; SLD, sobrevida livre de doença; UVB–NB UVB, UVB de faixa estreita; RT, radioterapia; Gy, Gray; MF un., micose fungoide unilesional.
Taxas de resposta e duração das respostas para os tratamentos sistêmicos para a MF/SS
| Tratamento | Estágio | Eficáciarespostaglobal (%) | Eficáciarespostacompleta (%) | Duração daresposta | Referências |
|---|---|---|---|---|---|
| Isotretinoína | MF/SS | 43–80 | 8–33 | DR: 3–15 meses | 22, 23 |
| Acitretina | Ia–IV | 59–64 | 4–34,6 | DR: 28 meses | 22–24 |
| Bexaroteno | I–IIa | 54–67 | 7–27 | DR: 9,8–17,2 meses | 23, 25 |
| ≥IIb | 45–54 | 2–13 | 23, 25 | ||
| Alfainterferona | Ia–IVa | 29–80 | 4–67 | DR: 5–8 meses | 26, 29 |
| Alfainterferona peguilada | Ib–III | 50–83 | 33–67 | – | 28 |
| Doxorrubicina lipossomal peguilada | Ib–IV | 41–88 | 6 | DR: 6–13,3 meses | 32, 33 |
| Gencitabina | T3–T4 | 47–70 | 11,5–22 | DR: 10–15 meses | 34, 38 |
| Clorambucila | SS | 85 | 35 | DR: 16,5 meses | 36 |
| III–IVb | 100 | 53,8 | 35 | ||
| Metotrexato, baixa dose | T2 | 33 | 33 | DR: 15 meses | 39 |
| III | 56 | 41 | DR: 31 meses | 39 | |
| Pralatrexato | IIa–IV | 40,8 | 6,1 | DR: 6 meses | 40 |
| Romidepsina | IIb | 34–38 | 6–7 | DR: 15 meses | 41 |
| Vorinostate | Ib–IVa | 30 | – | DR: 5 meses | 42 |
| Alentuzumabe | MF av.–SS | 38–84 | 0–47 | DR: 4 meses | 44, 45 |
| Brentuximabe vedotina | Ia–IVb | 50 | 10 | TPT: 13,4 meses | 46 |
| Mogamulizumabe | Ia–IVb/SS | 28 (MF: 21; SS: 37) | – | SLP: 7,7 meses | 47 |
| CHOP | IIb | 66 | – | TPT: 5,7 meses | 38 |
| EPOCH | IIb–IV | 80 | – | SLP: 8 meses | 38 |
| Fludarabina+CFM | IIb–III | 55 | – | DR: 10 meses | 38 |
| Fludarabina+IFN | IIa–IVb | 40–58 | – | SLP: 5,9 meses | 38 |
| IFN+PUVA | Ia–IVa | 80,6 | 75 | DR: 32 meses | 30 |
| – | – | SLP, 5 anos: 75% | 30 | ||
| Ia–IIa | 98 | 84 | SLP, 5 anos: 27% | 31 | |
| IFN peg+PUVA | Ia–IV | 88 | 44 | SLP: 30,9 meses | 27 |
| IFN+retinoide | Ia–IIb | 38 | 6 | – | 29 |
| Bexaroteno+PUVA | Ib–IIa | 77 | 31 | DR: 5,8 meses | 52 |
| FEC isolada | III–IVa1 | 24 | 9 | TPT: 14 meses | 49‐51 |
| FEC+IFN | III–IVa1 | 45,4 | 18,2 | – | 49, 50 |
| FEC+IFN+bexaroteno | III–IVa1 | 88,2 | 32,4 | DR: 4 meses | 49, 50 |
SLP, sobrevida livre de progressão; DR, duração da resposta; TPT, tempo para o próximo tratamento; MF av., micose fungoide avançada; SS, síndrome de Sézary; CHOP, ciclofosfamida, doxorrubicina, vincristina e prednisolona; EPOCH, etoposídeo, vincristina, doxorrubicina, ciclofosfamida e prednisolona; FEC, fotoférese extracorpórea; CFM, ciclofosfamida; IFN, interferona; IFN peg, interferona peguilada.
As modalidades de tratamento podem ser divididas em dois grupos: terapias direcionadas à pele e terapias sistêmicas. Os tratamentos podem ser utilizados como monoterapia ou terapia combinada. As terapias direcionadas à pele incluem agentes tópicos, fototerapia e radioterapia.7 As terapias sistêmicas incluem modificadores da resposta biológica, imunoterapias e agentes quimioterápicos.8 Para a MF em estágio inicial (estágio Ia–IIa), as terapias direcionadas à pele são o tratamento preferido, com o objetivo de controlar a doença, melhorar os sintomas e a qualidade de vida. Na MF em estágio inicial refratária, na MF em estágio tardio (estágio ≥ IIb) e na SS, é necessário o tratamento sistêmico, combinado ou não a uma terapia dirigida à pele. As modalidades de tratamento são revisadas neste artigo.
A aprovação e disponibilidade das drogas nos Estados Unidos, Europa e Brasil estão descritos na tabelas 6 e 7. A fotoférese extracorpórea, a fototerapia com UVB de faixa estreita, a fotoquimioterapia tipo PUVA e a radioterapia estão mais amplamente disponíveis.
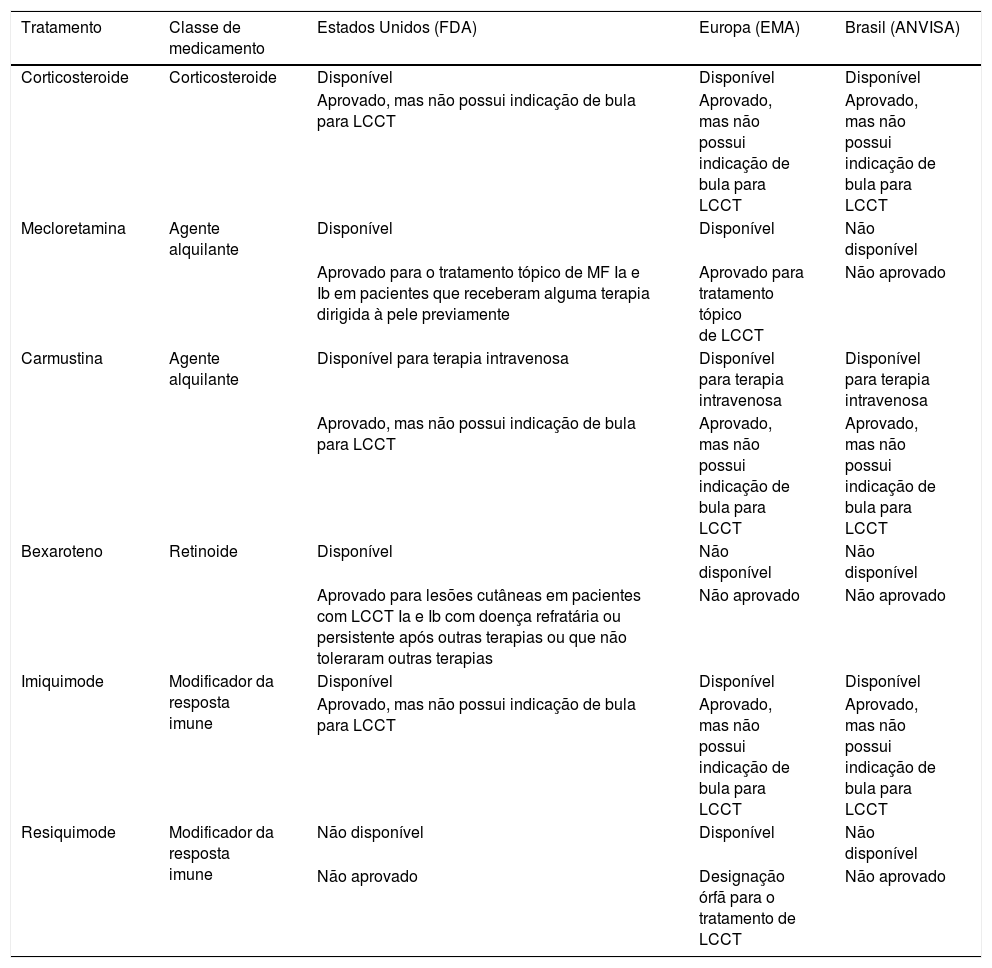
Aprovação de tratamentos tópicos para linfomas cutâneos de células T nos Estados Unidos, Europa e Brasil, de acordo com registro de bula nos órgãos reguladores locais (FDA, EMA e ANVISA), respectivamente
| Tratamento | Classe de medicamento | Estados Unidos (FDA) | Europa (EMA) | Brasil (ANVISA) |
|---|---|---|---|---|
| Corticosteroide | Corticosteroide | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Mecloretamina | Agente alquilante | Disponível | Disponível | Não disponível |
| Aprovado para o tratamento tópico de MF Ia e Ib em pacientes que receberam alguma terapia dirigida à pele previamente | Aprovado para tratamento tópico de LCCT | Não aprovado | ||
| Carmustina | Agente alquilante | Disponível para terapia intravenosa | Disponível para terapia intravenosa | Disponível para terapia intravenosa |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Bexaroteno | Retinoide | Disponível | Não disponível | Não disponível |
| Aprovado para lesões cutâneas em pacientes com LCCT Ia e Ib com doença refratária ou persistente após outras terapias ou que não toleraram outras terapias | Não aprovado | Não aprovado | ||
| Imiquimode | Modificador da resposta imune | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Resiquimode | Modificador da resposta imune | Não disponível | Disponível | Não disponível |
| Não aprovado | Designação órfã para o tratamento de LCCT | Não aprovado |
FDA, Food and Drug Administration (https://www.fda.gov/drugs); EMA, European Medical Agency (https://www.ema.europa.eu/en); ANVISA, Agência Nacional de Vigilância Sanitária (http://portal.anvisa.gov.br/); LCCT, linfomas cutâneos de células T; MF, micose fungoide.
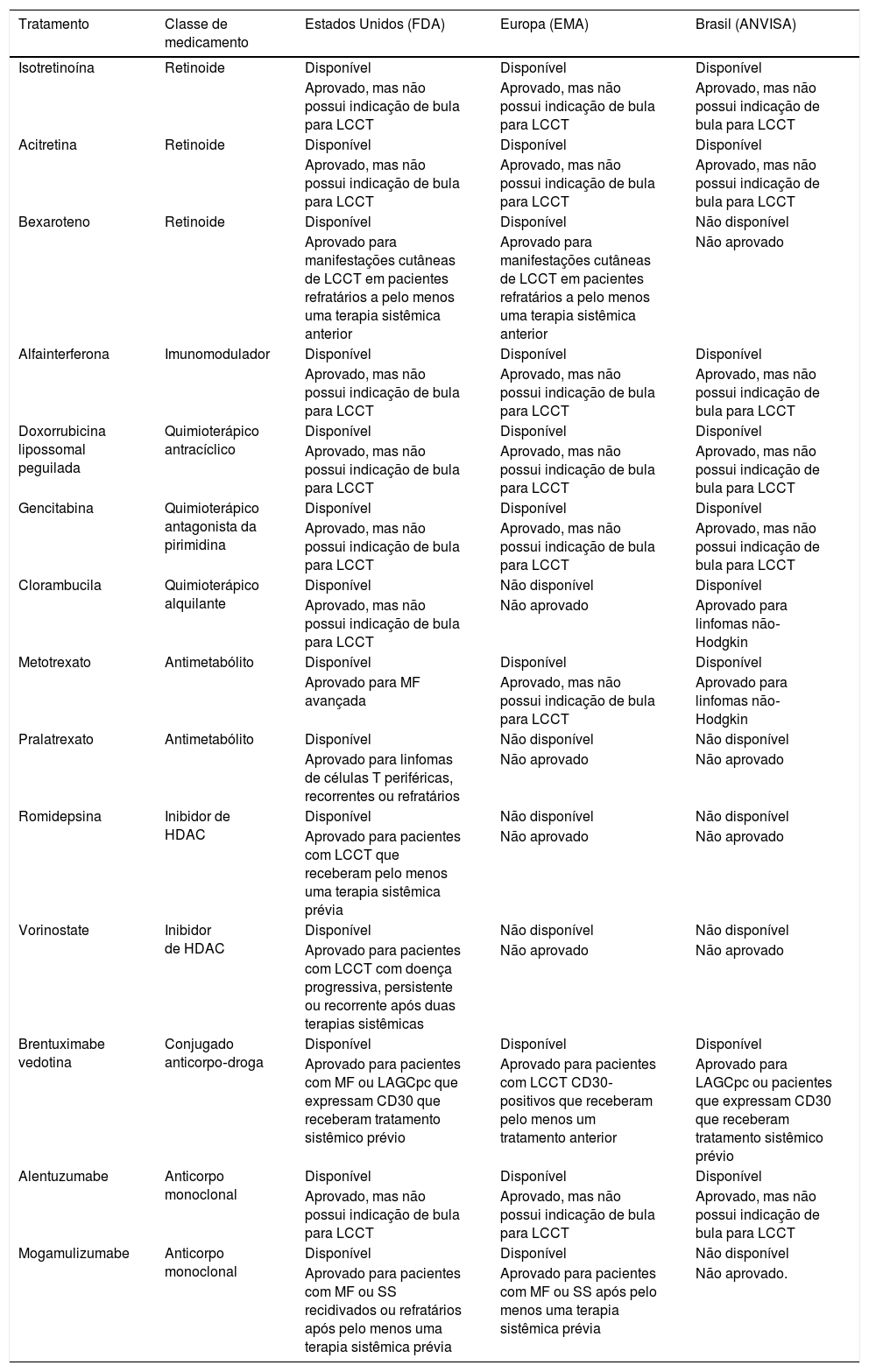
Aprovação de tratamentos sistêmicos para linfomas cutâneos de células T nos Estados Unidos, Europa e Brasil, de acordo com o registro de bula nas agências reguladoras locais FDA, EMA e ANVISA, respectivamente
| Tratamento | Classe de medicamento | Estados Unidos (FDA) | Europa (EMA) | Brasil (ANVISA) |
|---|---|---|---|---|
| Isotretinoína | Retinoide | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Acitretina | Retinoide | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Bexaroteno | Retinoide | Disponível | Disponível | Não disponível |
| Aprovado para manifestações cutâneas de LCCT em pacientes refratários a pelo menos uma terapia sistêmica anterior | Aprovado para manifestações cutâneas de LCCT em pacientes refratários a pelo menos uma terapia sistêmica anterior | Não aprovado | ||
| Alfainterferona | Imunomodulador | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Doxorrubicina lipossomal peguilada | Quimioterápico antracíclico | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Gencitabina | Quimioterápico antagonista da pirimidina | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Clorambucila | Quimioterápico alquilante | Disponível | Não disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Não aprovado | Aprovado para linfomas não‐Hodgkin | ||
| Metotrexato | Antimetabólito | Disponível | Disponível | Disponível |
| Aprovado para MF avançada | Aprovado, mas não possui indicação de bula para LCCT | Aprovado para linfomas não‐Hodgkin | ||
| Pralatrexato | Antimetabólito | Disponível | Não disponível | Não disponível |
| Aprovado para linfomas de células T periféricas, recorrentes ou refratários | Não aprovado | Não aprovado | ||
| Romidepsina | Inibidor de HDAC | Disponível | Não disponível | Não disponível |
| Aprovado para pacientes com LCCT que receberam pelo menos uma terapia sistêmica prévia | Não aprovado | Não aprovado | ||
| Vorinostate | Inibidor de HDAC | Disponível | Não disponível | Não disponível |
| Aprovado para pacientes com LCCT com doença progressiva, persistente ou recorrente após duas terapias sistêmicas | Não aprovado | Não aprovado | ||
| Brentuximabe vedotina | Conjugado anticorpo‐droga | Disponível | Disponível | Disponível |
| Aprovado para pacientes com MF ou LAGCpc que expressam CD30 que receberam tratamento sistêmico prévio | Aprovado para pacientes com LCCT CD30‐positivos que receberam pelo menos um tratamento anterior | Aprovado para LAGCpc ou pacientes que expressam CD30 que receberam tratamento sistêmico prévio | ||
| Alentuzumabe | Anticorpo monoclonal | Disponível | Disponível | Disponível |
| Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | Aprovado, mas não possui indicação de bula para LCCT | ||
| Mogamulizumabe | Anticorpo monoclonal | Disponível | Disponível | Não disponível |
| Aprovado para pacientes com MF ou SS recidivados ou refratários após pelo menos uma terapia sistêmica prévia | Aprovado para pacientes com MF ou SS após pelo menos uma terapia sistêmica prévia | Não aprovado. |
FDA, Food and Drug Administration (https://www.fda.gov/drugs); EMA, European Medical Agency (https://www.ema.europa.eu/en); ANVISA, Agência Nacional de Vigilância Sanitária (http://portal.anvisa.gov.br/); LCCT, linfomas cutâneos de células T; MF, micose fungoide; HDAC, histona desacetilase; LAGCpc, linfoma anaplásico de grandes células primário cutâneo; SS, síndrome de Sézary.
Os corticosteroides tópicos são os agentes anti‐inflamatórios mais utilizados na dermatologia. Inibem a ligação de linfócitos ao endotélio, inibem a adesão intercelular e induzem a apoptose de células linfoides neoplásicas. São frequentemente formulados como creme, pomada, loção ou gel. De acordo com o corticosteroide tópico e a formulação, a potência do medicamento pode ser estratificada em sete classes: classe I (super potente – p.ex., creme de propionato de clobetasol 0,05%), classe II (potente – p. ex., pomada de dipropionato de betametasona 0,05%), classe III (potência média superior – p. ex., creme de dipropionato de betametasona 0,05%), classe IV (potência média – p. ex., creme de acetonido de triamcinolona 0,1%), classe V (potência média inferior – p. ex., creme de valerato de betametasona 0,1%), classe VI (potência leve – p. ex., creme de desonida a 0,05%) e classe VII (menos potente – p. ex., creme de acetato de hidrocortisona a 1%). Os esteroides tópicos são aplicados uma ou duas vezes ao dia, por semanas a meses, até regressão completa ou melhora considerável das lesões. O uso prolongado de corticosteroides tópicos pode resultar em atrofia, estrias e irritação discreta da pele. Absorção sistêmica com o uso de corticosteroides de alta potência aplicados em grandes superfícies da pele, causando insuficiência adrenal clinicamente evidente e síndrome de Cushing, raramente são observadas.7,9
MecloretaminaA mecloretamina, clormetina ou mustarda nitrogenada é um agente quimioterápico alquilante que afeta células em rápida divisão, atuando como um agente citotóxico no DNA. A mustarda nitrogenada esteve originalmente disponível em pó liofilizado. O pó podia ser composto como creme ou solução aquosa. O creme era preparado na concentração inicial de 10 a 20mg de mustarda nitrogenada por 100g de petrolato ou similar. Em relação à preparação aquosa, os pacientes preparavam a solução na concentração de 10 a 20 mg/100 mL de água. As preparações eram aplicadas uma vez ao dia em lesões específicas ou na superfície total da pele (dependendo da classificação‐T), exceto na área genital, por semanas a meses. Atualmente, está disponível nos Estados Unidos e Europa como uma formulação em gel a 0,02% para aplicações mais localizadas. Eventos adversos de curto prazo incluem prurido, sensação de queimação e dermatite de contato irritante ou alérgica. No longo prazo, pode se observar hiper ou hipopigmentação da pele e um aumento discreto do risco de câncer da pele não‐melanoma, especialmente quando o uso da mostarda for combinado com outras terapias, como a fototerapia. O uso tópico da substância não causa mielossupressão.7,10,11
CarmustinaA carmustina, também conhecida como biscloretilnitrosoureia (BCNU), é um agente alquilante, que forma ligações cruzadas no DNA, impedindo a replicação e transcrição do DNA. Para preparar a solução alcoólica, 100 mg de carmustina em pó são dissolvidos em 5 mL de etanol a 95%. Essa solução de 5mL é então colocada em um recipiente de vidro e diluída em mais 50 mL de etanol a 95%. Isso produz uma concentração de 2mg/mL (0,2%), denominada solução estoque ou solução‐mãe. Para aplicações corporais totais, 5mL (10mg de BCNU) da solução‐mãe são diluídos em 60mL de água. A aplicação é feita uma vez por dia. Para lesões localizadas, o volume da solução é ajustado à área de envolvimento da pele. Para doenças extremamente limitadas (< 3% da pele envolvida), podem ser feitas aplicações com a solução‐mãe não diluída. A carmustina tópica também pode ser incorporada em uma pomada à base de petrolato a uma concentração de 10 mg/100g de petrolato. Geralmente, a dosagem diária máxima é de 10mg/dia, com cursos de tratamento de 6 a 12 semanas. Esse tratamento não costuma ser recomendado para pacientes com envolvimento cutâneo superior a 3%, pois a absorção sistêmica pode levar a toxicidades hematológicas. As toxicidades hematológicas incluem leucopenia, trombocitopenia e anemia. Reações locais são comumente observadas, e a maioria dos pacientes desenvolve eritema e sensação de queimação, principalmente nas dobras cutâneas.7,12
BexarotenoO bexaroteno é um retinoide (derivado da vitamina A) que ativa seletivamente os receptores de retinoides X, induzindo a diferenciação e apoptose das células. O bexaroteno tópico está disponível em uma formulação a 1%. O medicamento é aplicado nas lesões todas as noites durante a primeira semana de tratamento e, em seguida, duas vezes ao dia. O uso do bexaroteno geralmente é limitado a pacientes com envolvimento de menos de 15% da área de superfície corporal. Os efeitos adversos mais comuns são dermatite irritativa leve a moderada, prurido e queimação no local da aplicação.7,13
Imiquimode e resiquimodeO imiquimode é um agonista do receptor Toll‐like 7 que potencializa a produção de interferona. A interferona leva a efeitos antivirais, antiproliferativos e antiangiogênicos. Também estimula as células de Langerhans a migrarem para os linfonodos, ativando os linfócitos T. O imiquimode tópico está disponível como creme a 5% e é aplicado nas lesões, três a cinco vezes por semana, até a resolução. A toxicidade mais relatada é uma reação inflamatória local, com eritema, edema, vesículas e ulceração/erosão. Esses sinais refletem a ativação do sistema imunológico e, se nenhuma resposta inflamatória for observada, é menos provável que ocorra uma resposta adequada à terapia. Alguns pacientes podem apresentar sintomas sistêmicos (p. ex., sintomas gripais).7,14 O resiquimode é um agonista dos receptores Toll‐like 7 e 8. O receptor tipo Toll 8 é expresso por células dendríticas derivadas de células mieloides, e essas células são fortemente ativadas pela droga. Como o imiquimode, como agonista do receptor Toll‐like 7, a produção de interferona é aumentada. O medicamento, em gel de 0,06% ou 0,03%, é aplicado nas lesões da pele três vezes por semana, durante oito semanas. Observa‐se menor irritação da pele em relação ao imiquimode, com eritema e erosões superficiais. Pode ocorrer febre baixa por curto período. Não estão descritos eventos adversos graves.7,15
Terapias com radiaçãoFototerapiaÉ uma terapia física baseada em luz ultravioleta (UV). A luz UV atua causando parada do ciclo celular ou apoptose de queratinócitos, de células de Langerhans e de linfócitos e diminuindo seletivamente a produção de citocinas pró‐inflamatórias pelas células T. Apresenta diferentes modalidades de tratamento com base no comprimento de onda e na associação ou não a um psoraleno (agente fotossensibilizador). Os mais utilizados são a fototerapia com UVB de faixa estreita (NB‐UVB) e a PUVA (psoraleno+ultravioleta A), também conhecida como fotoquimioterapia. É uma opção de tratamento importante para pacientes com MF em estágio inicial ou como terapia adjuvante para estágios mais avançados. A dose e a duração do tratamento variam entre as instituições. Assim como o uso ou não de uma terapia de manutenção após a melhora das lesões (sendo usada com menos frequência para reduzir a dose acumulada total de UV).7,16
UVB de faixa estreitaA fototerapia com NB‐UVB compreende o comprimento de onda de 311 a 313 nm. Devido ao seu menor comprimento de onda, é absorvido principalmente na epiderme com menos capacidade de penetrar mais profundamente quando comparado ao UVA. Portanto, seus principais efeitos diretos são nos queratinócitos epidérmicos, nas células de Langerhans, no infundíbulo folicular e nas células da derme superior, incluindo linfócitos. É usado com mais frequência na doença em estágio de lesões não infiltradas (patches) (T1a e T2a). Apresenta vantagens sobre o PUVA por não requerer o uso de um agente fotossensibilizante (psoraleno), com menor risco de câncer de pele (com doses cumulativas) e pode ser utilizado na gravidez. O tratamento é administrado ambulatorialmente, três a cinco vezes por semana, com incremento gradual da dose. Os eventos adversos agudos mais comuns (em 24 horas) são eritema, prurido, queimação, formação de bolhas, bronzeamento e xerose. Em relação às neoplasias cutâneas, uma revisão de literatura publicada em 2005 que incluiu mais de 3.400 pacientes (principalmente pacientes com psoríase) tratados com UVB de banda larga (BB‐UVB) ou NB‐UVB, não mostrou aumento no risco de câncer de pele, exceto naqueles tratados com UVB e PUVA. A exposição crônica à luz UV está relacionada à formação de catarata e pterígio ocular. O uso de proteção ocular durante a fototerapia com UVB é indicado para evitar esse risco. No entanto, para aqueles que necessitam de tratamento periocular é seguro realizar as sessões com os olhos fechados, uma vez que há apenas uma transmissão desprezível de luz UV pelas pálpebras.7,16
Fotoquimioterapia PUVANesta modalidade de tratamento, agentes fotossensibilizantes (psoralenos) são utilizados por via oral ou tópica antes da exposição ao UVA (comprimento de onda de 320 a 400nm). O UVA (especialmente o UVA1 – 340 a 400nm) pode penetrar em toda a derme, podendo afetar os linfócitos da derme profunda, os fibroblastos, as células dendríticas dérmicas, os mastócitos, as células endoteliais, os macrófagos e partes mais profundas do folículo piloso. É uma opção melhor para o tratamento de lesões mais infiltradas em placas e da MF foliculotrópica. O tratamento com PUVA é administrado três vezes por semana até que a regressão das lesões seja alcançada. A proteção ocular deve ser usada não apenas durante as sessões, mas por 24 horas após a ingestão do psoraleno. Deve‐se notar que doses cumulativas de UV estão associadas a um risco aumentado de malignidades cutâneas associadas, principalmente carcinoma espinocelular, mas também carcinoma basocelular. Ainda há controvérsias quanto ao aumento do risco de melanoma. O psoraleno oral pode causar náusea e, com pouca frequência, hepatotoxicidade. Os psoralenos são absorvidos pelo cristalino, mas se difundem em 24 horas sem exposição ao UVA. Na presença de UVA, o 8‐MOP se liga a ácidos nucleicos e proteínas no cristalino e permanece por longos períodos, aumentando, teoricamente, o risco de catarata. Portanto, recomenda‐se usar proteção ocular UVA, durante a exposição à luz solar, após a ingestão de psoraleno por 24 horas.7,16
RadioterapiaA radioterapia (RT) envolve o uso de radiação ionizante por meio de fótons ou elétrons para destruir as células neoplásicas. A radiação ionizante atua causando danos ao DNA do tecido neoplásico, levando à morte celular. A MF é um tumor muito radiossensível, e a radioterapia é uma modalidade de tratamento muito eficaz.7
Radioterapia localPara lesões individuais ou localizadas, utiliza‐se a radioterapia convencional com fótons ou a radioterapia com feixe de elétrons.6,7 Regimes de baixa dose com 8Gy em duas frações são eficazes para placas ou pequenos tumores. Para o tratamento de grandes áreas, devem ser consideradas doses menores por fração (20‐30 Gy em 10 a 15 frações). O volume alvo clínico da radioterapia é definido com uma margem de pelo menos 1cm ao redor da lesão. A doença nodal periférica e a doença visceral também podem ser tratadas com RT localizada. A MF é uma doença multifocal, e o controle local com radioterapia é frequentemente usado como abordagem paliativa. Portanto, recomenda‐se o uso da dose mínima de radioterapia para obter o controle local, o que possibilita retratamentos ou tratamentos em áreas adjacentes. O evento adverso observado na radioterapia localizada é a radiodermatite; pode ocorrer fenômeno de recidiva de radiodermatite em pacientes tratados com quimioterapia.17–19
Terapia com feixe de elétrons em toda peleA terapia com feixe de elétrons de toda a pele (total skin electron beam therapy – TSEBT) é uma técnica especial que propicia uma irradiação homogênea de toda a superfície cutânea. A dose total habitual para tratamento é de 36Gy, administrada com 1‐2Gy por sessão durante cinco a nove semanas.20 As taxas de remissão completa são altas, entretanto são frequentes as recidivas. O TSEBT em dose baixa é realizado com 12Gy, 1Gy por fração durante três semanas.21 Diretrizes de consenso sobre o uso do TSEBT no MF estão publicadas.18 A toxicidade relacionada ao tratamento depende da dose de radiação usada e da localização do tumor. Os eventos adversos incluem eritema, perda de pelos, suspensão temporária de crescimento das unhas, edema de mãos e pés, hemorragias nasais menores, bolhas nos dedos das mãos e pés, anidrose, parotidite leve, ginecomastia em homens, ceratite pelo uso dos protetores oculares internos, distrofia ungueal permanente, xerose, alopecia permanente, disestesia nas extremidades dos dedos.7 A TSEBT em dose baixa é mais bem tolerada e os eventos adversos são transitórios e mais leves em comparação com a dose padrão. Entretanto as taxas de resposta são inferiores.21
Terapias sistêmicasRetinoides: isotretinoina, acitretina e bexarotenoRetinoides são análogos naturais e sintéticos da vitamina A que se ligam a várias classes de proteínas, incluindo proteínas de ligação a retinoides e receptores nucleares de retinoides. Levam à ativação de regiões reguladoras de DNA envolvidas na regulação do crescimento, diferenciação e apoptose das células. Os receptores nucleares de retinoides pertencem a duas famílias: os receptores de ácido retinoico (RAR) e os receptores retinoides X (RXR). A isotretinoína é um retinoide não aromático de primeira geração. Foi o primeiro retinoide usado para o tratamento off label de LCCT. É utilizado por via oral em doses diárias de 0,2 a 1,0 mg/kg. Os eventos adversos incluem secura da pele e mucosas, elevação de lipídios no sangue e teratogenicidade (a gravidez deve ser evitada por um mês após a descontinuação do tratamento).22 A acitretina é um retinoide monoaromático de segunda geração. Sua estrutura o torna mais lipofílico com maior biodisponibilidade que os retinoides de primeira geração. É utilizado por via oral em doses diárias de 0,3‐0,5 mg/kg. O perfil de segurança é semelhante ao da isotretinoína, mas a gravidez deve ser evitada por 2‐3 anos após a descontinuação do tratamento.22 O bexaroteno é um retinoide poliaromático de terceira geração que é altamente seletivo para o receptor RXR. É administrado por via oral em uma dose diária de 300mg/m2. O perfil de segurança é semelhante ao da isotretinoína e acitretina, mas com eventos adversos adicionais como hipotireoidismo central e hipertrigliceridemia na maioria dos pacientes. A gravidez deve ser evitada por um mês após a descontinuação do tratamento.8,23–25
InterferonaA interferona (IFN) é uma citocina Th1. Existem três tipos de interferonas recombinantes (alfa, beta, gama) atualmente disponíveis para uso terapêutico. A IFN‐α existe na forma peguilada. Todas as IFN recombinantes são ativas no tratamento da MF/SS, mas a IFN‐α é a mais estudada. Ela regula o ciclo celular, promove a supressão de oncogenes e modula a adesão celular.26 A IFN‐γ é mais comumente estudada e utilizada no Japão. O tratamento com a IFN‐α é geralmente iniciado com uma dose de 3 milhões de unidades por via subcutânea, três vezes por semana. A dosagem pode ser aumentada até 9 milhões de unidades por aplicação. A IFN‐α peguilada, formulação na qual o fármaco é encapsulado nos lipossomas, resultando em meia‐vida aumentada e melhor acúmulo nos tecidos tumorais, é administrada por via subcutânea na dose de 1,5μg/kg semanalmente.27,28 A IFN‐γ é utilizado por via subcutânea na dose de 50μg/m2 para os pacientes com área de superfície corporal maior do que 0,5 m2 e de 1,5μg/kg/dose para pacientes com área de superfície corporal igual ou inferior a 0,5 m2. É usado diariamente ou três vezes por semana.29 Os eventos adversos das interferonas são dependentes da dose e incluem sintomas semelhantes aos da gripe, disfunção tireoidiana, aumento de transaminases, leucopenia, trombocitopenia, depressão e arritmias.29–31
Doxorrubicina lipossomal peguiladaA doxorrubicina é um agente citotóxico antracíclico. Liga‐se aos ácidos nucleicos, inibindo a progressão da topoisomerase II, interrompendo o processo de replicação do DNA. A doxorrubicina lipossomal peguilada é encapsulada nos lipossomas, resultando em meia‐vida aumentada e melhor acúmulo nos tecidos tumorais. A doxorrubicina lipossomal peguilada é administrada por via intravenosa, na dose de 20mg/m2 nos dias 1 e 15, a cada 28 dias. Os principais eventos adversos são geralmente leves ou moderados e incluem anemia, astenia, náusea, vômito e eritrodisestesia palmoplantar.8,32,33
GencitabinaA gencitabina (2’, 2’‐difluorodeoxicitidina) é um agente quimioterápico clássico da família dos análogos de nucleosídeos. Atua bloqueando a formação de novo DNA, resultando em morte celular. É utilizada em doses de 1.200mg/m2 por via intravenosa nos dias 1, 8 e 15 de um regime de 28 dias. O tratamento é bem tolerado e o principal evento adverso é a toxicidade hematológica, geralmente leve.8,34
ClorambucilaA clorambucila é um agente alquilante e forma ligações cruzadas no DNA, causando danos ao DNA e afetando a replicação e transcrição. A clorambucila pode ser usado por via oral em regime contínuo com 2‐6 mg/dia de clorambucila associada a 20 mg/dia de prednisona, ou em pulsos de 10‐12mg/dia de clorambucila por três dias e fluocortolona 75 mg no primeiro dia, 50 mg no segundo dia e 25 mg no terceiro dia) a cada duas semanas.35,36 As doses e intervalos podem ser reduzidos e prolongados com a melhora da doença. O uso contínuo e prolongado da clorambucila aumenta o risco de mielossupressão e desenvolvimento de leucemia, especialmente leucemia mieloide aguda.
Quimioterapia combinadaAs combinações mais usadas são: ciclofosfamida, doxorrubicina, vincristina e prednisona (CHOP); ciclofosfamida, vincristina e prednisona (CVP); ciclofosfamida, doxorrubicina, vincristina e etoposídeo (CHOEP); CVP mais metotrexato (MTX). Os regimes de quimioterapia combinados estão associados com altas taxas de resposta (70%‐80%), mas frequentemente de curta duração (em torno de quatro meses). A quimioterapia multidroga também é associada a mielossupressão e complicações infecciosas. Portanto, com raras exceções, a administração sequencial de uma única substância quimioterápica é a estratégia preferida.37,38
Substâncias antifolato: metotrexato e palatrexatoO MTX é um antimetabólito do tipo antifolato. Inibe competitivamente dihidrofolato redutase e consequentemente o metabolismo do ácido fólico, atuando pela inibição da síntese de DNA, RNA, timidilatos e proteínas. O MTX em doses baixas tem sido usado para tratar a MF em estágio inicial e a SS por muitos anos.39 O MTX é usado na dose de 10 a 25 mg, uma vez por semana, via oral ou subcutânea. Os efeitos adversos incluem sintomas gastrintestinais (náuseas, vômito, estomatite, diarreia), anemia, leucopenia, trombocitopenia, aumento de transaminases, fibrose hepática e pneumonite.8,40 O palatrexato também é um análogo do folato com atividade demonstrada em pacientes com MF/SS. É administrado por via intravenosa na dosagem de 15 mg/m2 por semana durante três semanas, em ciclos de quatro semanas. Os eventos adversos são semelhantes aos observados com o MTX, mas tendem a ser mais comuns e graves. Os efeitos colaterais mais frequentes incluem mucosite, fadiga, náusea, vômito, anorexia, toxicidade cutânea, epistaxe e anemia.8,40
Inibidores da histona‐desacetilase: romidepsina e vorinostateA histona‐desacetilase (HDAC) é uma classe de enzimas responsáveis por catalisar a remoção de grupos acetil das histonas, sendo moduladores cruciais da regulação epigenética da transcrição. Duas substâncias dessa classe têm a aprovação da FDA para LCCT, principalmente em casos refratários: romidepsina e vorinostate.41,42 A romidepsina é administrada como um agente único na dose de 14mg/m2 por via intravenosa nos dias 1, 8 e 15 de um ciclo de 28 dias. Os eventos adversos relacionados mais comuns são náuseas e fadiga.43 O vorinostate é um inibidor da HDAC por via oral. É administrado em dose diária de 400 mg por dia. Se não for tolerado, pode ser reduzida para 300mg por dia ou 300mg, cinco dias por semana. Os eventos adversos mais comuns são diarreia, fadiga, náusea e anorexia.43
AlentuzumabeO alentuzumabe é um anticorpo monoclonal humanizado anti‐CD52. O CD52 é uma glicoproteína de superfície presente nos linfócitos T e B, monócitos e macrófagos. O alentuzumabe se liga ao CD52, causando destruição das células neoplásicas por citotoxicidade celular dependente de anticorpos e fixação do complemento. As taxas de resposta são mais altas na SS do que na MF porque o alentuzumabe leva ao esgotamento das células T de memória central no sangue e na pele dos pacientes com SS. Por outro lado, o medicamento não afeta as células T efetoras de memória residentes na pele na MF. Evidências recentes sugerem que o alentuzumabe não é eficaz na MF tumoral ou transformada. A dosagem padrão é de 30mg por via intravenosa, três vezes por semana, por até 12 semanas.8,44 O alentuzumabe subcutâneo também pode ser utilizado em regime de doses baixas de 10 a 15 mg em dias alternados, três vezes por semana.45 Reações infusionais (febre, náuseas, hipotensão, fadiga, erupção cutânea, urticária, broncoespasmo) são observadas em mais da metade dos pacientes. Citopenias (linfopenia, neutropenia, anemia, trombocitopenia) são observadas em quase todos os pacientes. Infecções graves (citomegalovírus, herpes simples generalizado, aspergilose fatal e pneumonia por micobactérias), podem ser observadas, em especial em pacientes intensamente pré‐tratados.8 O regime de dose baixa pode reduzir as complicações infecciosas em pacientes com MF.45 O tratamento com alentuzumabe requer profilaxia antibiótica e antiviral, bem como observação cuidadosa para o desenvolvimento de infecção cardíaca e toxicidade.8
Brentuximabe vedotinaA brentuximabe vedotina (BV) é um conjugado anticorpo‐droga direcionado à molécula CD30. O CD30 pertence à superfamília do receptor do fator de necrose tumoral. A droga ligada ao anticorpo é a monometil auristatina E (MMAE), um agente antitubulina. Em tecidos normais, o CD30 tem um perfil de expressão bastante restrito às células T, B e NK/T ativadas, mas é altamente expresso nas células de Reed‐Sternberg do linfoma de Hodgkin, no linfoma anaplásico de grandes células e em outros subtipos de linfomas não‐Hodgkin, como no linfoma anaplásico de grandes células primário cutâneo (LAGCpc) e em determinados casos de MF. Esse perfil de expressão torna o CD30 um alvo ideal para terapias baseadas em anticorpos monoclonais. Após a ligação do BV ao CD30 na superfície das células neoplásicas, e sua internalização, ocorre a liberação da MMAE. A MMAE vai, então, exercer seu potente efeito citostático, inibindo a montagem dos microtúbulos, induzindo a parada do ciclo celular, e resultando na morte por apoptose das células tumorais. A segurança e eficácia da BV foi avaliada no primeiro estudo de fase III contra uma terapia padrão (MTX ou bexaroteno) para LCCT, tendo demonstrado altas taxas de respostas duráveis e clinicamente significantes. A dose inicial típica é de 1,8 mg/kg administrada por via intravenosa a cada três semanas. As principais toxicidades consistem em neuropatia periférica, neutropenia, fadiga, náusea e alopecia.46
MogamulizumabeO mogamulizumabe é um anticorpo monoclonal humanizado que tem como alvo o receptor de quimiocina CC4 (CCR4) expresso nas células neoplásicas. O CCR4 é expresso na superfície das células tumorais da maioria dos pacientes com leucemia‐linfoma de células T do adulto (ATLL) e é seletivamente expresso em outros subtipos de linfoma de células T periféricas e linfoma de células T cutâneo. Após a ligação ao seu alvo, o mogamulizumabe atua por citotoxicidade mediada por células dependente de anticorpos para destruir as células tumorais. O mogamulizumabe é administrado por via intravenosa na dose de 1,0 mg/kg semanalmente por quatro semanas, seguido de uma dose a cada duas semanas até a progressão da doença. Erupções relacionadas à infusão e erupções cutâneas decorrentes do uso do fármaco são comuns; outras toxicidades são diarreia, náusea, trombocitopenia, disgeusia e elevação de creatinina sérica.47
Fotoférese extracorpóreaA fotoférese extracorpórea (FEC) é uma terapia imunomoduladora na qual a leucaférese remove os leucócitos do paciente, que são então tratados com 8‐metoxipsoraleno e UVA e reinfundidos no paciente. É o tratamento de primeira linha para SS e MF eritrodérmica.6,48 Cada ciclo da FEC é geralmente administrado em dois dias consecutivos a cada duas a quatro semanas por pelo menos seis meses. Com o desenvolvimento do 8‐MOP em solução, a FEC pode ser administrada diretamente na bolsa de aférese sanguínea, e o paciente não é exposto à absorção sistêmica da substância, minimizando os efeitos adversos gastrintestinais e a fotossensibilidade sistêmica. Os efeitos colaterais esporádicos incluem dor de cabeça, pirexia, mialgia, anemia leve, trombocitopenia e fotofobia. Hipotensão, síncope vasovagal, infecção no local da injeção, agravamento das lesões cutâneas e sepse, em geral decorrentes do acesso venoso central, são raras. Não há relatos de infecções oportunistas ou malignidades, pois a FEC não é um tratamento imunossupressor.48–51
Terapias combinadasA combinação de tratamentos é uma estratégia bem estabelecida para aumentar a eficácia terapêutica nos tratamentos para MF. Inclui a combinação de terapias direcionadas à pele com terapias sistêmicas ou de duas ou mais terapias sistêmicas. As combinações mais utilizadas são retinoides e PUVA (RE‐PUVA), interferona e PUVA ou FEC com interferona e/ou bexaroteno.49,52
Transplante de células‐tronco hematopoiéticas (TCTH)O transplante alogênico de células‐tronco (TCTH‐alo) pode induzir remissões duráveis e é o único tratamento com intenção curativa. Por causa das altas taxas de morbimortalidade associadas ao TCTH‐alo, uma boa seleção de pacientes é essencial para o sucesso do tratamento. Há a necessidade de aconselhamento cuidadoso e a indicação deve se concentrar, principalmente, em pacientes mais jovens e com boa performance, com estágios avançados da doença e baixa carga tumoral no momento do transplante e que estejam cientes do alto risco de progressão e mau prognóstico.53
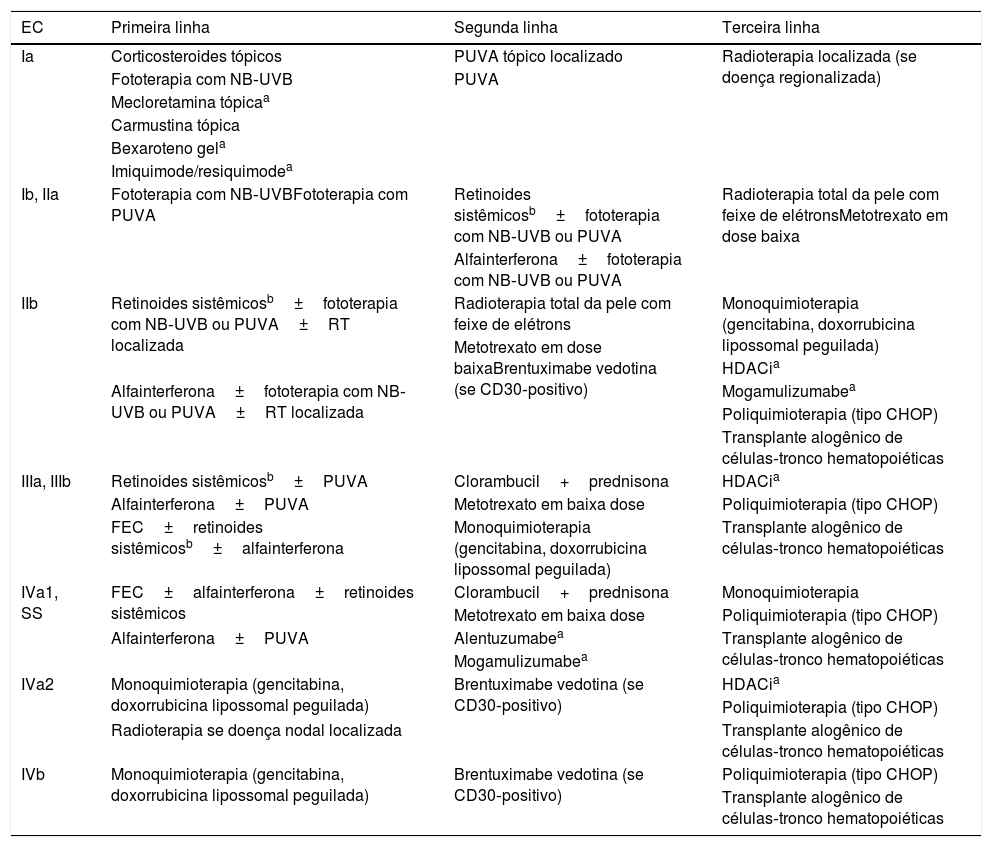
Taxas de respostas e recomendações de tratamentoAs taxas e duração das respostas com os diversos tratamentos dirigidos à pele, tratamentos sistêmicos e combinação de tratamentos reportados na literatura encontram‐se nas tabelas 4 e 5. Importante ressaltar que os estudos são metodologicamente heterogêneos e muitos não são controlados. O resumo das recomendações de tratamento do Grupo de Linfoma Cutâneos das Divisões de Dermatologia e Hematologia do HC‐FMUSP pode ser visto na tabela 8. As recomendações levam em consideração a experiência clínica e a melhor evidência científica para os tratamentos em diversos estágios da doença. Na doença precoce refratária e na doença avançada, a despeito da utilização de terapias sistêmicas (modificadores da resposta biológica e/ou quimioterapia), é frequente a associação das terapias direcionadas à pele. A disponibilidade e/ou aprovação no Brasil, para os tratamentos descritos, não garantem acesso aos medicamentos pelos pacientes tanto nos setores públicos quanto nos privados. Vários fatores podem influenciar no acesso aos tratamentos, como protocolos clínicos institucionais, diretrizes estaduais, protocolos de tratamento da saúde suplementar, particularidades de planos de saúde/seguros‐saúde, pareceres de comissões de padronização de medicamentos ou mesmo as solicitações individuais de medicamentos por pacientes. Existem drogas tanto para tratamentos dirigidos à pele quanto para tratamentos sistêmicos que são de fundamental importância no tratamento da MF e SS, mas que ainda não estão disponíveis no Brasil. Desse modo, orienta‐se que as recomendações sejam avaliadas e adaptadas à realidade de cada serviço. Acreditamos que o desenvolvimento de um Protocolo Clínico Nacional para a Abordagem dos Linfomas Cutâneos seja fundamental para garantir acesso universal de todos os pacientes aos diversos tratamentos disponíveis no país. Inquestionavelmente, os melhores cuidados administrados aos pacientes oncológicos traduzem‐se em diminuição dos impactos físicos e emocionais relacionados à própria doença e ao indivíduo, assim como em uma significativa redução dos custos econômicos associados, diretos e indiretos.
Recomendações do Grupo de Linfomas Cutâneos das Divisões de Dermatologia e Hematologia do HC‐FMUSP para o tratamento da Micose fungoide e síndrome de Sézary
| EC | Primeira linha | Segunda linha | Terceira linha |
|---|---|---|---|
| Ia | Corticosteroides tópicos | PUVA tópico localizado | Radioterapia localizada (se doença regionalizada) |
| Fototerapia com NB‐UVB | PUVA | ||
| Mecloretamina tópicaa | |||
| Carmustina tópica | |||
| Bexaroteno gela | |||
| Imiquimode/resiquimodea | |||
| Ib, IIa | Fototerapia com NB‐UVBFototerapia com PUVA | Retinoides sistêmicosb±fototerapia com NB‐UVB ou PUVA | Radioterapia total da pele com feixe de elétronsMetotrexato em dose baixa |
| Alfainterferona±fototerapia com NB‐UVB ou PUVA | |||
| IIb | Retinoides sistêmicosb±fototerapia com NB‐UVB ou PUVA±RT localizada | Radioterapia total da pele com feixe de elétrons | Monoquimioterapia (gencitabina, doxorrubicina lipossomal peguilada) |
| Metotrexato em dose baixaBrentuximabe vedotina (se CD30‐positivo) | |||
| HDACia | |||
| Alfainterferona±fototerapia com NB‐UVB ou PUVA±RT localizada | Mogamulizumabea | ||
| Poliquimioterapia (tipo CHOP) | |||
| Transplante alogênico de células‐tronco hematopoiéticas | |||
| IIIa, IIIb | Retinoides sistêmicosb±PUVA | Clorambucil+prednisona | HDACia |
| Alfainterferona±PUVA | Metotrexato em baixa dose | Poliquimioterapia (tipo CHOP) | |
| FEC±retinoides sistêmicosb±alfainterferona | Monoquimioterapia (gencitabina, doxorrubicina lipossomal peguilada) | Transplante alogênico de células‐tronco hematopoiéticas | |
| IVa1, SS | FEC±alfainterferona±retinoides sistêmicos | Clorambucil+prednisona | Monoquimioterapia |
| Metotrexato em baixa dose | Poliquimioterapia (tipo CHOP) | ||
| Alfainterferona±PUVA | Alentuzumabea | Transplante alogênico de células‐tronco hematopoiéticas | |
| Mogamulizumabea | |||
| IVa2 | Monoquimioterapia (gencitabina, doxorrubicina lipossomal peguilada) | Brentuximabe vedotina (se CD30‐positivo) | HDACia |
| Poliquimioterapia (tipo CHOP) | |||
| Radioterapia se doença nodal localizada | Transplante alogênico de células‐tronco hematopoiéticas | ||
| IVb | Monoquimioterapia (gencitabina, doxorrubicina lipossomal peguilada) | Brentuximabe vedotina (se CD30‐positivo) | Poliquimioterapia (tipo CHOP) |
| Transplante alogênico de células‐tronco hematopoiéticas |
HC‐FMUSP, Hospital de Clínicas da Faculdade de Medicina da Universidade de São Paulo; EC, estágio clínico; PUVA, psoraleno+ultravioleta A; NB‐UVB, UVB de faixa estreita; RT, radioterapia; HDACi, inibidores da histona‐desacetilase; CHOP, ciclofosfamida, doxorrubicina, vincristina e prednisolona; SS, síndrome de Sézary; FEC, fotoférese extracorpórea.
Como a maioria dos tratamentos disponíveis para MF e SS raramente induzem longos períodos de remissão ou cura completa, os principais objetivos do tratamento são controlar os sintomas da doença, melhorar a qualidade de vida do paciente, prolongar a sobrevida livre de progressão e a sobrevida global. Além disso, como uma doença indolente, com uma sobrevida em cinco anos relacionada à doença de cerca de 90% na maioria dos casos, e com a importância do microambiente tumoral no controle da progressão da doença, deve‐se inicialmente escolher terapias que possam ser utilizadas a longo prazo, adiando o uso de terapias sistêmicas agressivas, como a quimioterapia com múltiplas drogas, reservadas apenas para raras ocasiões. Nos estágios iniciais da doença, as terapias direcionadas à pele constituem o padrão de cuidado. Na doença avançada ou refratária são utilizadas as terapias sistêmicas isoladas ou combinadas com as terapias dirigidas à pele, a despeito das raras taxas observadas de remissão completa e dos poucos estudos controlados que as referendam. O papel do TCTH‐alo ainda não está bem definido, mas pode ser útil em alguns pacientes adequadamente selecionados. A disponibilidade de tratamento também é bastante heterogênea dentro de um país, assim como entre os diferentes países.54 Espera‐se que o esforço realizado por centros especializados em todo o mundo, com o objetivo de condução de estudos multicêntricos e multidisciplinares internacionais, proporcione uma melhor compreensão da doença, o desenvolvimento de novos tratamentos e um padrão de atendimento mais eficiente e uniforme aos portadores de MF e SS.
Suporte financeiroNenhum.
Contribuição dos autoresJosé Antonio Sanches: Concepção e elaboração do manuscrito.
Jade Cury‐Martins: Concepção e elaboração do manuscrito.
Rodrigo Martins Abreu: Concepção e elaboração do manuscrito.
Denis Miyashiro: Interpretação dos dados e na revisão crítica do manuscrito.
Juliana Pereira: Interpretação dos dados e na revisão crítica do manuscrito.
Todos os autores aprovaram a versão final do manuscrito e concordam em ser responsáveis por todos os aspectos do trabalho, garantindo que as questões relacionadas à precisão ou integridade de qualquer parte do trabalho sejam investigadas e resolvidas adequadamente.
Conflito de interessesCury‐Martins J e Pereira J foram sub‐investigadoras em um estudo financiado pela Takeda e recebem honorários da Takeda para palestras ocasionais. Miyashiro D não tem nada a declarar. Abreu RM é gerente médico científico em onco‐hematologia da Takeda. Sanches JA foi o principal investigador de um estudo financiado pela Takeda.
Como citar este artigo: Sanches JA, Cury‐Martins J, Abreu RM, Miyashiro D, Pereira J. Mycosis fungoides and Sézary syndrome: focus on the current treatment scenario. An Bras Dermatol. 2021;96:458–71.
Trabalho realizado na Divisão de Clínica Dermatológica, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil.
