





A via de sinalização Janus Quinase (JAK) e transdutor de sinal/ativador da transcrição (STAT) medeia importantes processos celulares, como a resposta imune, carcinogênese, diferenciação, divisão e morte celular. Portanto, medicamentos que interfiram nos diferentes padrões de sinalização JAK‐STAT têm potencial indicação para várias condições médicas. Os principais alvos dermatológicos dos inibidores da via JAK‐STAT são doenças inflamatórias ou autoimunes como psoríase, vitiligo, dermatite atópica e alopecia areata, porém, diversas dermatoses estão em investigação para ampliar esse rol de indicações. Como os inibidores da via JAK‐STAT devem, paulatinamente, ocupar espaço relevante na prescrição dermatológica, esta revisão apresenta os principais medicamentos disponíveis, seus efeitos imunológicos e suas características farmacológicas relacionadas à eficácia clínica e de segurança, a fim de substanciar a melhor prática dermatológica.
A via de sinalização Janus Quinase (JAK) e transdutor de sinal/ativador da transcrição (STAT) compreende uma família de moléculas ligadas aos domínios intracelulares dos receptores de diversas citocinas e fatores de crescimento, que medeiam sua sinalização até o núcleo.1 A denominação dessas quinases como Janus refere‐se ao deus romano que representava o início ou abertura de um processo. Janus tinha duas faces, que estão associadas aos dois domínios das JAK: um catalítico e um semelhante à quinase. Historicamente, o início do dia, do mês e do ano era destinado em sacramento a Janus; assim, segundo a lenda, o mês de janeiro origina‐se de seu nome.
A via JAK‐STAT pertence a um complexo sistema de proteína quinases, conservado evolutivamente, em que mediadores extracelulares controlam a expressão de genes específicos envolvidos em diversas funções celulares como mitose, diferenciação, apoptose, hematopoese, desenvolvimento do sistema imune (inato e adaptativo) e atividade de glândulas exócrinas. Além disso, participa de respostas celulares a agressões como hipoxia, irradiação ultravioleta, estímulo por endotoxinas, estresse oxidativo e hiperosmolar.2
Desse modo, diversas dermatoses inflamatórias ou autoimunes são alvo de estudo para a indicação de inibidores da via JAK‐STAT (iJAK).3 Uma vez que essa classe terapêutica se revelou promissora em substituir o uso crônico dos imunossupressores clássicos (p. ex., ciclosporina, azatioprina, micofenolato, metotrexato, corticosteroides) em virtude de sua seletividade de ação, os iJAK devem, paulatinamente, ocupar espaço na prescrição dermatológica.
Esta é uma revisão narrativa que englobou artigos publicados sobre iJAK com direcionamento para as principais doenças dermatológicas nas bases de dados MEDLINE, SciELO e Google Scholar, que avaliou o período de 1992 (dos primeiros fundamentos do processo de sinalização intracelular) até os dias atuais. Apresentamos os principais medicamentos disponíveis, seus efeitos imunológicos e suas características farmacológicas relacionadas à eficácia clínica e de segurança, a fim de substanciar a melhor prática dermatológica.
Via JAK‐STATAs enzimas JAK são tirosina quinases agregadas aos domínios intracelulares dos receptores transmembrana de certas citocinas e fatores de crescimento. Após o acoplamento das moléculas nos domínios extracelulares de seus receptores ocorre modificação conformacional em sua estrutura, que leva à fosforilação de resíduos específicos de tirosina nos dímeros de JAK. Essa fosforilação possibilita o recrutamento de proteínas como os fatores de transcrição STAT, que se dimerizam e são translocadas para o núcleo das células (via Ran de importação nuclear) a fim de regular a transcrição de genes específicos (fig. 1).4
 de sinalização JAK‐STAT. (A) Diversas citocinas e fatores de crescimento presentes no meio extracelular dependem de receptores transmembrana para dar início ao processo de sinalização celular e transcrição nuclear dos genes associados a cada via. (B) O acoplamento da citocina com o domínio extracelular do receptor transmembrana leva a mudança em sua conformação e fosforilação dos dímeros de JAK, situados no domínio intracelular do receptor, e que transfosforilam seus resíduos terminais de tirosina. Esse processo induz a dimerização e fosforilação de unidades inativas de STAT, que migram para o núcleo e medeiam a transcrição de genes relacionados à via da citocina específica. (C) Inibidores da via de sinalização JAK‐STAT (iJAK) impedem a fosforilação da JAK, interrompendo a sinalização nuclear da citocina ou fator de crescimento. Fonte: os autores")
Representação esquemática da via (canônica) de sinalização JAK‐STAT. (A) Diversas citocinas e fatores de crescimento presentes no meio extracelular dependem de receptores transmembrana para dar início ao processo de sinalização celular e transcrição nuclear dos genes associados a cada via. (B) O acoplamento da citocina com o domínio extracelular do receptor transmembrana leva a mudança em sua conformação e fosforilação dos dímeros de JAK, situados no domínio intracelular do receptor, e que transfosforilam seus resíduos terminais de tirosina. Esse processo induz a dimerização e fosforilação de unidades inativas de STAT, que migram para o núcleo e medeiam a transcrição de genes relacionados à via da citocina específica. (C) Inibidores da via de sinalização JAK‐STAT (iJAK) impedem a fosforilação da JAK, interrompendo a sinalização nuclear da citocina ou fator de crescimento. Fonte: os autores
Cada subtipo de JAK contém um resíduo de tirosina específico para a fosforilação ATP‐dependente. Essa especificidade as diferencia entre si e entre as mais de 500 tirosinas quinases humanas.5
Os principais componentes da via JAK‐STAT constituem quatro enzimas JAK (JAK1, JAK2, JAK3 e tirosina quinase 2 [Tyk2]), e sete enzimas STAT (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B e STAT6). Elas se distribuem como dímeros cuja composição varia de acordo com o tipo de receptor transmembrana e o tipo celular envolvido.1 Logo, de acordo com o padrão de inibição para diferentes JAK‐STAT, diferentes perfis de sinalização de citocina são afetados.
O bloqueio seletivo da sinalização de grupos de citocinas é uma característica do efeito imunomodulador dos iJAK em comparação com os imunossupressores convencionais (p. ex., ciclosporina ou corticosteroides), que suprimem uma vasta gama de mediadores; ou com os imunobiológicos, que suprimem, especificamente, uma citocina.6 A tabela 1 apresenta as principais citocinas e os fatores de crescimento afetados pelo bloqueio dos subtipos de JAK.
Principais citocinas e fatores de crescimento influenciados pela sinalização de diferentes subtipos de JAK, em humanos
| JAK | Citocinas e fatores de crescimento |
|---|---|
| JAK1 | IFN‐α, IFN‐β, IFN‐γ, IL‐2, IL‐4, IL‐6, IL‐7, IL‐9, IL‐10, IL‐11, IL‐13, IL‐15, IL‐19, IL‐21, IL‐22, IL‐27, IL‐28, IL‐29, IL‐31, IL‐35, fator neurotrófico ciliar (CNTF), oncostatina‐M (OSM), cardiotrofina (CT‐1) |
| JAK2 | IFN‐γ, IL‐3, IL‐5, IL‐6, IL‐10, IL‐11, IL‐13, IL‐12, IL‐19, IL‐22, IL‐23, IL‐27, IL‐35, fator de estimulação de colônias de granulócitos e macrófagos (GM‐CSF), eritropoitina (EPO), tireoide peroxidase (TPO), leptina, oncogene viral mieloproliferativo da leucemia (mpl), prolactina, hormônio do crescimento (GH) |
| JAK3 | IL‐2, IL‐4, IL‐7, IL‐9, IL‐13, IL‐15, IL‐21 |
| TYK2 | IFN‐α, IFN‐β, IL‐10, IL‐11, IL‐12, IL‐19, IL‐22, IL‐23, IL‐27, IL‐28, IL‐29 |
Como as unidades JAK estão acopladas apenas a receptores de citocinas tipos I e II, os iJAK não medeiam, diretamente, a sinalização de TNF (α, β), TGF (α, β), EGF, quimiocinas (p. ex., IL‐8, CXC, CX3C), PDGF e IL‐1 (α, β), que são patogênicas em uma série de dermatoses.7,8
A via de sinalização JAK‐STAT constitui profícua linha de pesquisa em biologia celular envolvendo aspectos ligados ao comportamento de variantes mutantes de suas enzimas, a regulação intracelular por inibidores/ativadores, efeitos da ativação não canônica e interação com outras vias inflamatórias, apoptóticas ou de crescimento celular que são assuntos clinicamente relevantes, porém que ultrapassam o escopo deste texto.9–16
Principais características dos iJAK na dermatologiaOs iJAK são moléculas pequenas (< 500 kD), o que favorece sua absorção intestinal e permeação cutânea, apesar de serem pouco lipofílicas (log p entre 1,5 e 2). Por constituírem medicamentos finais e não dependerem de metabolização para o efeito farmacológico, sua rápida absorção leva a efeito clínico precoce; porquanto, seus picos plasmáticos e meia‐vidas de eliminação também são breves (< 18 horas).17
Nas formulações tópicas, iJAK não implicam em níveis séricos elevados dos medicamentos, minimizando efeitos adversos (EA) sistêmicos e interações medicamentosas.17,18 Além disso, atingem alta concentração na epiderme e derme superior, o que justifica sua eficácia em eczemas, psoríase e vitiligo, conforme discutido adiante. Entretanto, recomenda‐se não ultrapassar 20% da superfície cutânea, utilizar camada fina e não prolongar seu uso, com o intuito de reduzir a chance de efeitos sistêmicos.17
Quanto à seletividade, os iJAK podem ser classificados como de primeira geração (p. ex., tofacitinibe, baricitinibe) ou não seletivos, ou de segunda geração (p. ex., upadacitinibe, abrocitinibe, ritlecitinibe), considerados os mais seletivos para um dos subtipos de JAK. Além disso, os iJAK diferem quanto ao bloqueio enzimático (reversível ou covalente) e o sítio de ligação (tipo I, II e alostérico).19
Quando consideradas suas estruturas moleculares, os iJAK podem ser agrupados como iJAKα (baricitinibe, delgocitinibe, ruxolitinibe, tofacitinibe) ou iJAKβ (abrocitinibe, upadacitinibe, filgotinibe, deucravacitinibe). Os iJAKα têm estrutura semelhante à purina e sistema bicíclico condensado formado por heterociclos de pirimidina/pirrol. Todas essas substâncias exibem um grupo cianeto nucleofílico para o sítio de ligação da quinase. Dada a semelhança dos segmentos JH1 entre as isoformas JAK, os iJAKα as inibem todas, apesar de diferentes afinidades. Os iJAKβ foram especificamente sintetizados por modelagem química para otimizar as ligações, na tentativa de combinar a estrutura dos iJAKβ com um respectivo sítio de ligação, gerando maior seletividade. Porém, os iJAKβ são menos comparáveis entre si quanto à estrutura química.20
Em virtude da semelhança estrutural das enzimas do grupo JAK, os diferentes iJAK exercem algum efeito inibitório em todos os seus quatro subtipos, de acordo com a concentração utilizada. A tabela 2 resume as características farmacológicas dos principais iJAK de uso atual na dermatologia. Entretanto, a maior parte dos indicadores farmacológicos e farmacodinâmicos é estimada por ensaios em que as concentrações inibitórias são realizadas por testes in vitro que empregam diferentes metodologias, sem considerar variações metabólicas ligadas a sexo, faixa etária, ascendência étnica, composição corporal, comorbidades e uso de medicamentos concomitantes. Essas características justificam cautela em sua prescrição e seguimento de longo prazo, uma vez que os resultados de farmacovigilância das principais substâncias ainda estão em andamento em virtude da recente aprovação (menos de cinco anos) pelas agências regulatórias internacionais.
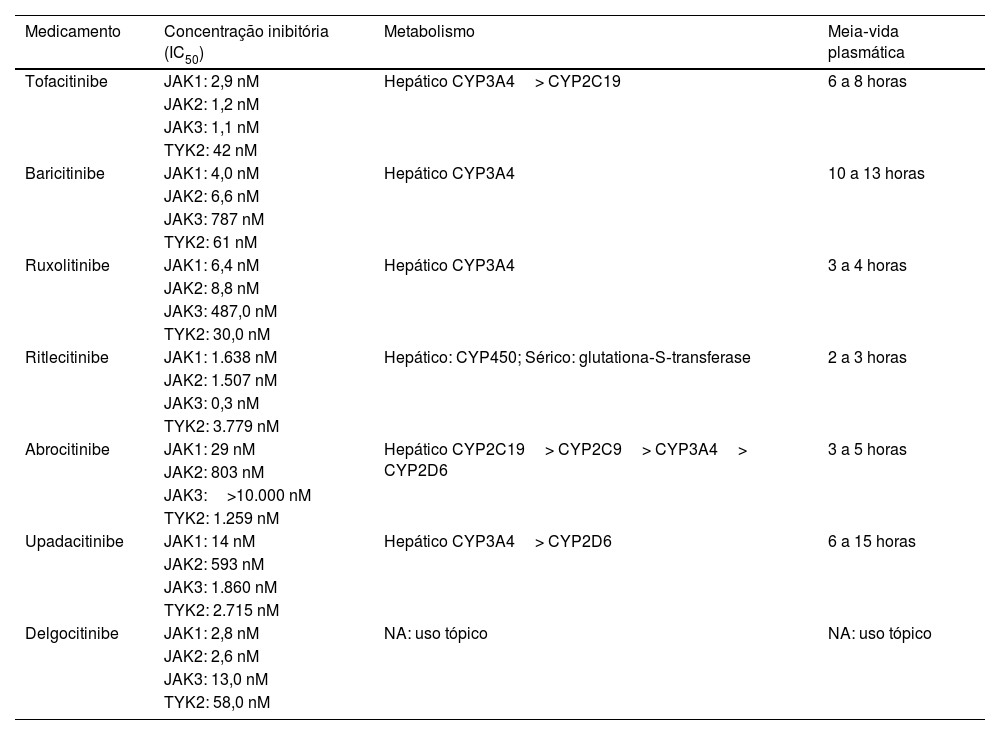
Características farmacológicas dos principais inibidores da via JAK‐STAT disponíveis no mercado
| Medicamento | Concentração inibitória (IC50) | Metabolismo | Meia‐vida plasmática |
|---|---|---|---|
| Tofacitinibe | JAK1: 2,9 nM | Hepático CYP3A4> CYP2C19 | 6 a 8 horas |
| JAK2: 1,2 nM | |||
| JAK3: 1,1 nM | |||
| TYK2: 42 nM | |||
| Baricitinibe | JAK1: 4,0 nM | Hepático CYP3A4 | 10 a 13 horas |
| JAK2: 6,6 nM | |||
| JAK3: 787 nM | |||
| TYK2: 61 nM | |||
| Ruxolitinibe | JAK1: 6,4 nM | Hepático CYP3A4 | 3 a 4 horas |
| JAK2: 8,8 nM | |||
| JAK3: 487,0 nM | |||
| TYK2: 30,0 nM | |||
| Ritlecitinibe | JAK1: 1.638 nM | Hepático: CYP450; Sérico: glutationa‐S‐transferase | 2 a 3 horas |
| JAK2: 1.507 nM | |||
| JAK3: 0,3 nM | |||
| TYK2: 3.779 nM | |||
| Abrocitinibe | JAK1: 29 nM | Hepático CYP2C19> CYP2C9> CYP3A4> CYP2D6 | 3 a 5 horas |
| JAK2: 803 nM | |||
| JAK3:>10.000 nM | |||
| TYK2: 1.259 nM | |||
| Upadacitinibe | JAK1: 14 nM | Hepático CYP3A4> CYP2D6 | 6 a 15 horas |
| JAK2: 593 nM | |||
| JAK3: 1.860 nM | |||
| TYK2: 2.715 nM | |||
| Delgocitinibe | JAK1: 2,8 nM | NA: uso tópico | NA: uso tópico |
| JAK2: 2,6 nM | |||
| JAK3: 13,0 nM | |||
| TYK2: 58,0 nM |
Tal como ocorre com outras medicações, exceto em reações imunologicamente mediadas, os EA e riscos ligados a medicamentos que interferem nas vias de sinalização e transcrição de citocinas, fatores de crescimento celular e indução da apoptose celular são diretamente proporcionais aos seguintes condicionantes: (i) ocorrência de doenças concomitantes (tuberculose latente, infecção por HIV, HTLV‐1, doença de Chagas, doenças autoimunes, doença intestinal inflamatória [DII], trombofilias, hepatopatias, nefropatias e hematológica); (ii) dose da medicação; (iii) longevidade do tratamento, e (iv) vias metabólicas alteradas por polimorfismo gênico (deficiência da glicose‐6‐fosfato desidrogenase, acetilador lento, HLA de predisposição a reações adversas graves como no caso do abacavir, HLA‐B*5701).21
Mesmo assim, muitos EA podem ser pouco frequentes, ou mesmo não estar presentes em estudos de fase III com limitado número de pacientes e curta exposição ao fármaco, que somente serão identificados pelos relatos de farmacovigilância, já que a maioria dos iJAK tem menos de cinco anos de comercialização.22 Cabe ao dermatologista estar atento à possibilidade de EA oriundos do uso prolongado desses fármacos, assim como identificar subgrupos de pacientes suscetíveis e, principalmente, a possibilidade de interações medicamentosas.
Interações medicamentosasO envelhecimento populacional e a disponibilidade de novos fármacos favorecem o uso concomitante de medicamentos pela população, o que maximiza a possibilidade de interações medicamentosas, por exemplo, via citocromo P450 (CYP).23
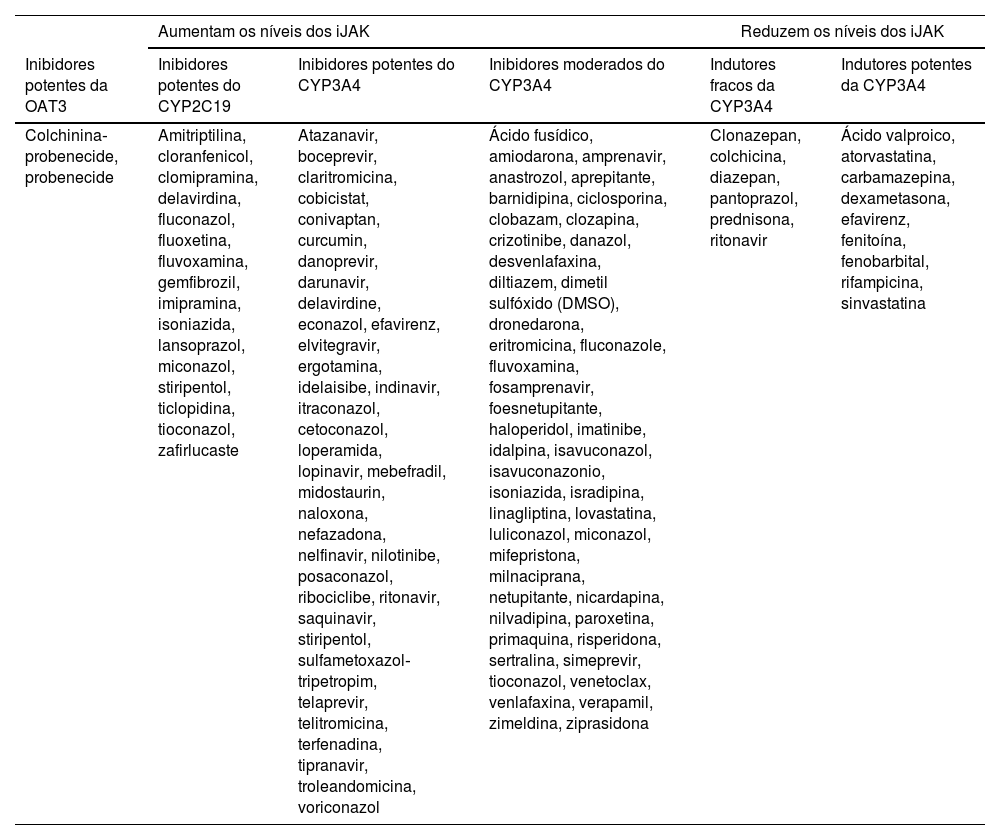
Em pacientes com artrite reumatoide (AR) que receberam iJAK, identificou‐se em mais de 10% deles a prescrição concomitante de medicamentos com potencial interação medicamentosa, como inibidores da bomba OAT3 (organic anion transporter 3), inibidores potentes do CYP3A4 e CYP2C19.23
Enquanto os iJAK são primariamente metabolizados no fígado pelo sistema CYP, via enzimas metabolizadoras do CYP3A4, a extensão do metabolismo por essas vias e outras enzimas varia de acordo com o iJAK utilizado, bem como seu nível de excreção renal. A tabela 3 fornece uma extensa lista de medicamentos que, potencialmente, interferem com CYP3A4, CYP2C19 e bomba OAT3 e que devem ser considerados no uso concomitante de iJAK.
Medicamentos que potencialmente interferem com os níveis séricos e segurança dos inibidores da via JAK‐STAT
| Aumentam os níveis dos iJAK | Reduzem os níveis dos iJAK | ||||
|---|---|---|---|---|---|
| Inibidores potentes da OAT3 | Inibidores potentes do CYP2C19 | Inibidores potentes do CYP3A4 | Inibidores moderados do CYP3A4 | Indutores fracos da CYP3A4 | Indutores potentes da CYP3A4 |
| Colchinina‐probenecide, probenecide | Amitriptilina, cloranfenicol, clomipramina, delavirdina, fluconazol, fluoxetina, fluvoxamina, gemfibrozil, imipramina, isoniazida, lansoprazol, miconazol, stiripentol, ticlopidina, tioconazol, zafirlucaste | Atazanavir, boceprevir, claritromicina, cobicistat, conivaptan, curcumin, danoprevir, darunavir, delavirdine, econazol, efavirenz, elvitegravir, ergotamina, idelaisibe, indinavir, itraconazol, cetoconazol, loperamida, lopinavir, mebefradil, midostaurin, naloxona, nefazadona, nelfinavir, nilotinibe, posaconazol, ribociclibe, ritonavir, saquinavir, stiripentol, sulfametoxazol‐tripetropim, telaprevir, telitromicina, terfenadina, tipranavir, troleandomicina, voriconazol | Ácido fusídico, amiodarona, amprenavir, anastrozol, aprepitante, barnidipina, ciclosporina, clobazam, clozapina, crizotinibe, danazol, desvenlafaxina, diltiazem, dimetil sulfóxido (DMSO), dronedarona, eritromicina, fluconazole, fluvoxamina, fosamprenavir, foesnetupitante, haloperidol, imatinibe, idalpina, isavuconazol, isavuconazonio, isoniazida, isradipina, linagliptina, lovastatina, luliconazol, miconazol, mifepristona, milnaciprana, netupitante, nicardapina, nilvadipina, paroxetina, primaquina, risperidona, sertralina, simeprevir, tioconazol, venetoclax, venlafaxina, verapamil, zimeldina, ziprasidona | Clonazepan, colchicina, diazepan, pantoprazol, prednisona, ritonavir | Ácido valproico, atorvastatina, carbamazepina, dexametasona, efavirenz, fenitoína, fenobarbital, rifampicina, sinvastatina |
Destaque‐se que fazem parte dessa lista medicamentos de uso corriqueiro, como ansiolíticos, antidepressivos, antifúngicos, antilipemiantes e anti‐hipertensivos, e os dermatologistas devem ponderar os riscos dessas interações. Inclusive, substâncias de uso recreativo (p. ex., Cannabis sp.) e extratos vegetais (p. ex., Echinacea purpurea, Hypericum perforatum), que são frequentemente omitidos pelos pacientes, têm efeito sobre a metabolização hepática de medicamentos.24,25 No entanto, o breve uso concomitante de iJAK com potenciais substâncias que promovem interação, como fluconazol para candidose vulvovaginal, não implica em prejuízo.
Diferentes iJAK de uso sistêmico têm particularidades farmacocinéticas e metabólicas dependentes de sua fração de ligação com proteínas plasmáticas, taxa de metabolização hepática e atividade dos seus metabólitos (tabela 2).
O tofacitinibe é rapidamente absorvido por via oral, com pico de concentração plasmática em 1 hora, atingindo equilíbrio (steady‐state) em até 48 horas. Sua biodisponibilidade oral é de 74%, independente da ingesta de alimentos, e 40% da substância se ligam a proteínas plasmáticas.20 É eliminado principalmente via hepática (70%) e renal (30%), o que demanda doses menores em hepatopatas e nefropatas. A principal via de metabolização utiliza o CYP3A4, com pequena contribuição do CYP2C19, além de ser substrato da glicoproteína‐P. Não foram demonstradas interações com midazolam, contraceptivos orais ou metformina.26
O upadacitinibe é absorvido em ampla faixa de pH (entre 1‐7,5) sem grande interferência alimentar, e apenas 52% se ligam às proteínas plasmáticas; portanto, não são esperadas interações relevantes dependentes do deslocamento proteico. Nenhum ajuste de dose é necessário para indivíduos com insuficiência renal ou hepática leve ou moderada.27 O upadacitinibe é metabolizado pela CYP3A4, com participação menor de CYP2D6. O uso concomitante com cetoconazol aumenta a concentração máxima (Cmáx) do medicamento e a curva de concentração plasmática (AUC) no tempo em 70% e 75%, respectivamente. O uso concomitante com rifampicina diminui a Cmáx e AUC em 50% e 60%, respectivamente. Não há interação medicamentosa com varfarina, omeprazol, midazolam e etinilestradiol+levonorgestrel.26
O abrocitinibe, após absorção, liga‐se em 37% às proteínas plasmáticas; a excreção é principalmente renal (85%) e fecal (9,5%). Quanto à forma não metabolizada, 0,6% são excretadas nos rins e 0,3% nas fezes.20 A coadministração deve seguir a seguintes recomendações: (i) uso concomitante com inibidores do CYP2C19 – a dose do abrocitinibe deve ser reduzida pela metade; (ii) uso concomitante com indutores potentes do CYP2C9/CYP2C19 – não recomendado; (iii) inibidores do OAT3 – sem necessidade de ajuste.28 Como exemplos, atenção especial: (i) fluconazol, fluoxetina, fluvoxamina e ticlopidina (inibidores do CYP2C19); (ii) apalutamida e rifampicina (uso não recomendado); e (iii) probenecida e teriflunomida (sem necessidade de ajuste de dose).28 Insuficiência hepática não tem efeito clinicamente relevante na farmacocinética e segurança do abrocitinibe, autorizando seu uso sem ajuste da dose em casos de insuficiência hepática leve ou moderada.29
O baricitinibe é rapidamente absorvido por via oral em jejum, com pico plasmático em 1 hora e biodisponibilidade de 80%. Alimentos gordurosos e calóricos diminuem sua absorção em 29%, atrasando o pico plasmático para 3 horas. Ocorre ligação de 50% às proteínas plasmáticas, e steady‐state é atingido em até 48 horas. Menos de 10% do baricitinibe ingerido sofrem metabolismo pelo CYP3A4, não se detectando metabólitos no plasma, o que dispensa ajuste de dose na insuficiência hepática leve ou moderada. O baricitinibe é excretado sem alteração na urina (69%) e nas fezes (15%), enquanto 6% da dose são excretados como metabólitos. A filtração glomerular é o principal mecanismo de excreção e secreção ativa pelo OAT3, glicoproteína‐P, BCRP e pela MATE2‐K. Assim, a concentração plasmática do baricitinibe aumenta em pacientes com disfunção renal leve (1,4×) e moderada (2,2×). Em pacientes com clearence de creatinina entre 30‐60mL/min, a dose do baricitinibe deve ser reduzida para 2mg/dia, e não é recomendado quando o clearance de creatinina for<30mL/min.30
O ajuste nas doses do tofacitinibe ou baricitinibe, mas não do upadacitinibe, é necessário com a progressão da gravidade da insuficiência renal. Embora a dose do tofacitinibe precise ser ajustada para pacientes com insuficiência hepática moderada, não é o caso do baricitinibe ou do upadacitinibe.31 Insuficiência renal moderada e grave levaram a maior exposição à porção ativa do abrocitinibe, sugerindo que a dose de abrocitinibe deva ser reduzida pela metade nesses casos.32
Infecções e eventos adversos geraisOs efeitos farmacológicos dos iJAK são proporcionais às doses utilizadas, de modo que sua prescrição deve levar em consideração a menor dose clinicamente eficaz, a fim de minimizar os riscos associados à imunossupressão ou à inibição da JAK2, associada aos EA hematológicos.
O uso de iJAK tópicos demonstrou poucos EA sistêmicos, porém acne, já relatada em pacientes com iJAK orais, foi observada em alguns relatos e séries de casos.3
A maioria dos eventos infecciosos relacionados aos iJAK é relatada pelo uso do tofacitinibe, liberado para comercialização em 2012, de modo que se torna evidente o maior tempo e o grande número de doentes expostos a essa medicação e, consequentemente, o maior número de publicações na literatura.33–36O estudo ORALSURV envolveu 4.362 indivíduos com AR, com mais de 50 anos e pelo menos um fator de risco cardiovascular. Doentes em uso prévio de metotrexato foram alocados para receber tofacitinibe 5mg, 2×/dia; tofacitinibe 10mg, 2×/dia; ou anti‐TNFα (etanercepte ou adalimumabe). Em relação às infecções, o estudo demonstrou que tofacitinibe conferia risco similar de infecções em relação aos anti‐TNFα, exceto pela propensão em reativar vírus latentes (p. ex., varicela‐zóster, herpes simplex e citomegalovírus).33
Com relação aos eventos infecciosos graves (EIG), o ORALSURV identificou risco semelhante para a dose de 5mg de tofacitinibe e os anti‐TNFα, mesmo em pacientes com mais de 65 anos.33 Os dados dos EIG no estudo foram consistentes com os dados dos programas de desenvolvimento dos iJAK e agentes biológicos modificadores do comportamento da AR (bDMARD) atualmente aprovados, nos quais as taxas de EIG foram semelhantes (3‐4 EIG/100 pacientes‐ano), com taxas mais elevadas em idosos.33,37
Os EAs mais comuns relatados nos estudos de fase III dos iJAK sistêmicos foram cefaleia, náuseas e nasofaringite. Infecções virais como herpes simples (HSV, herpes simplex virus), erupção variceliforme de Kaposi (eczema herpético [EH]) e herpes‐zóster (HZ) constituem os EA dermatológicos mais comuns em pacientes com dermatite atópica (DA) sob tratamento com iJAK.13 A maioria dos dados sobre infecção pelo HSV procede do uso do tofacitinibe na AR, artrite psoriásica e DII. No entanto, há menor incidência de infecção pelo HSV, HZ e EH nos ensaisos clínicos de iJAK1 (seletivos) em pacientes com DA.38,39
No estudo JADE REGIMEN foram incluídos 1.233 pacientes com DA moderada a grave em
fase aberta (12 semanas) em monoterapia com abrocitinibe 200mg/dia.39 Ao final de 12 semanas, 798 pacientes (64,7%) foram considerados respondedores ao medicamento e aleatoriamente alocados no estudo cego com abrocitinibe 200mg/dia, 100mg/dia ou placedo (40 semanas). Considerando a fase de estudo aberto (indução) e cego, os pacientes completaram 52 semanas de seguimento. Na fase de indução (12 semanas), foram observados EIGs em seis pacientes (0,5%), com HZ em nove pacientes (0,7%). Na fase cega (40 semanas), as taxas de EIG e HZ variaram entre os pacientes alocados nos três diferentes grupos: (i) placebo, 267 pacientes, foram observados EIG em dois casos (0,7%) e em HZ dois casos (0,7%); (ii) abrocitinibe 100mg/dia, 265 pacientes, EIG em dois casos (0,8%) e HZ em dois casos (0,8%); (iii) abrocitinibe 200mg/dia, 266 pacientes, foram observados EIG em cinco casos (1,9%), e HZ em nove casos (3,4%). Somando‐se os três grupos da fase cega (20 semanas; 798 pacientes), foram foram nove casos (1,1%) de EIG e 13 (1,6%) de HZ.39
Em metanálise sobre a eficácia e segurança de abrocitinibe, baricitinibe e upadacitinibe em pacientes com DA moderada a grave, os autores concluíram que quando se analisou o número total de EA (TEAE; do inglês, treatment‐emergent adverse events), o upadacitinibe e o abrocitinibe demonstraram maior incidência que o grupo placebo, e o abrocitinibe esteve associado a aumento nos TEAE, quando comparado com o baricitinibe. No entanto, quando se analisaram esses iJAK em relação aos TEAE e dose da medicação utilizada, apenas a dose de 30mg/dia do upadacitinibe aumentou a incidência dos TEAE: odds ratio (OR)=13,64. Ainda na metanálise de rede, a comparação dos eventos adversos de abrocitinibe versus placebo resultou em OR=5,57; baricitinibe versus placebo, OR=2,92; abrocitinibe versus baricitinibe, OR=1,92; abrocitinibe versus upadacitinibe, OR=0,41; e baricitinibe versus upadacitinibe, OR=0,21. Os autores da metanálise ressaltam que a maioria dos estudos de ensaios clínicos foi conduzida com avaliações de eficácia e segurança em um período curto de avaliação, sem comparação ainda em estudos entre eles, e que estudos de eficácia e segurança de longo prazo devem ser conduzidos.40
Como exemplos de estudos de fase III publicados com casuística considerável, randomizados e placebo‐controlados, Measure Up1 e Measure Up2 avaliaram, respectivamente, 847 e 836 pacientes com DA moderada a grave alocados em grupos recebendo 30mg/dia, 15mg/dia de upadacitinibe ou placebo. Os desfechos primários de eficácia foram avaliados na semana 16, e os EA foram registrados (tabela 4).
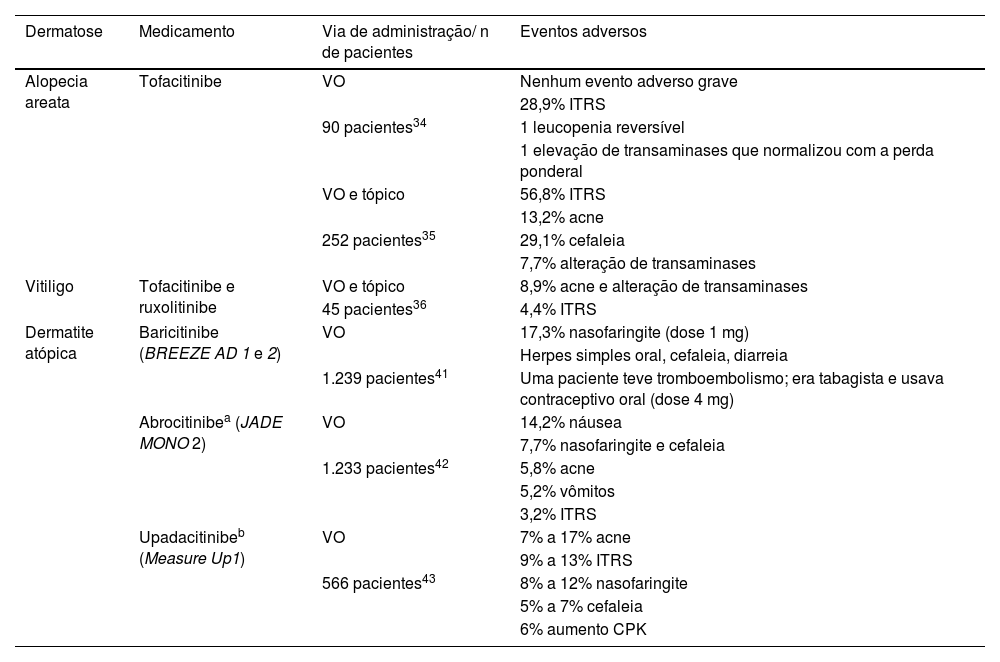
Principais eventos adversos por ordem de frequência das principais dermatoses em que os inibidores da via JAK‐STAT são utilizados
| Dermatose | Medicamento | Via de administração/ n de pacientes | Eventos adversos |
|---|---|---|---|
| Alopecia areata | Tofacitinibe | VO | Nenhum evento adverso grave |
| 28,9% ITRS | |||
| 90 pacientes34 | 1 leucopenia reversível | ||
| 1 elevação de transaminases que normalizou com a perda ponderal | |||
| VO e tópico | 56,8% ITRS | ||
| 13,2% acne | |||
| 252 pacientes35 | 29,1% cefaleia | ||
| 7,7% alteração de transaminases | |||
| Vitiligo | Tofacitinibe e ruxolitinibe | VO e tópico | 8,9% acne e alteração de transaminases |
| 45 pacientes36 | 4,4% ITRS | ||
| Dermatite atópica | Baricitinibe (BREEZE AD 1 e 2) | VO | 17,3% nasofaringite (dose 1 mg) |
| Herpes simples oral, cefaleia, diarreia | |||
| 1.239 pacientes41 | Uma paciente teve tromboembolismo; era tabagista e usava contraceptivo oral (dose 4 mg) | ||
| Abrocitinibea (JADE MONO 2) | VO | 14,2% náusea | |
| 7,7% nasofaringite e cefaleia | |||
| 1.233 pacientes42 | 5,8% acne | ||
| 5,2% vômitos | |||
| 3,2% ITRS | |||
| Upadacitinibeb (Measure Up1) | VO | 7% a 17% acne | |
| 9% a 13% ITRS | |||
| 566 pacientes43 | 8% a 12% nasofaringite | ||
| 5% a 7% cefaleia | |||
| 6% aumento CPK |
CPK, creatinofosfoquinase; ITRS, infecção do trato respiratório superior; VO, via oral.
No que se refere às complicações infectocontagiosas, é importante considerar que as prevalências das doenças variam com a epidemiologia de cada país. A via JAK‐STAT faz a transmissão de sinalização intracelular de mais de 50 citocinas e fatores de crescimento e é considerada a via de comunicação central do sistema imune, inclusive na defesa contra patógenos.
A alteração funcional do sistema JAK‐STAT pode ocorrer não apenas por ação farmacoterápica, mas também por mutações germinativas de perda ou ganho de função, as quais determinam diversos fenótipos imunes e doenças mieloproliferativas. Como exemplos: (i) mutações com perda de função da JAK3 são relacionadas com imunodeficiência combinada grave (SCID; do inglês, severe combined immunodeficiency); (ii) perda de função por mutações na STAT1 pode gerar deficiência de respostas ligadas às interferonas (IFN), tipo 1 (α e β) e IFN‐γ, ocasionando aumento na suscetibilidade de infecções virais; (iii) mutações com perda da função da JAK1, TYK2, STAT1 e STAT5B determinam infecções bacterianas intracelulares, e a deficiência da STATB5 pode ocasionar pneumonias de repetição; e (iv) mutação com ganho de função da STAT1 pode determinar infecções recorrentes por Candida spp., pois a STAT1 antagoniza os efeitos antifúngicos mediados pela IL‐17 via STAT3, suprimindo essa sinalização intracelular.44
Pacientes com dermatoses crônicas inflamatórias apresentam maior risco de infecção em decorrência tanto de sua própria doença (p. ex., disbiose cutânea com colonização de estafilococos na DA) quanto pelo tratamento ao qual são submetidos, em particular imunossupressores como ciclosporina, azatioprina, micofenolato e metotrexato.44 Como exemplo, na AR há o dobro de risco de esses pacientes apresentarem infecções graves, particularmente broncopulmonares e geniturinárias, que contribuem para maior mortalidade.45 Assim, ao prescrever iJAK, um dos fatores a se considerar são os potenciais EAs dessas substâncias, mas também a idade do paciente (imunossenescência), comorbidades associadas e aspectos da patogênese da doença.
Apesar de as infecções oportunistas se mostrarem mais frequentes em todas as classes de iJAK, não foram observadas diferenças em relação ao risco de EIGs em uma recente análise de rede de ensaios clínicos.45 Contudo, os ensaios clínicos individuais não foram desenvolvidos para examinar desfechos raros.
Uma análise dos estudos de extensão de longo prazo (EELP) sobre o tofacitinibe encontrou incidência de EIGs de 2,7 casos por 100 pacientes‐ano.46 Quando os iJAK foram associados a drogas antirreumáticas modificadoras da doença (DMARDs; do inglês, disease‐modifying antirheumatic drugs) em um registro nos EUA com 21.832 pacientes com AR, houve maior ocorrência de EIGs com tofacitinibe associado a outro DMARD na taxa de 3,67 vs. 2,01 EIG para cada 100 pacientes‐ano entre os pacientes em uso apenas do DMARD. A incidência de EIG na associação tofacitinibe+DMARD foi, inclusive, maior que nos pacientes que utilizaram tofacitinibe+anti‐TNFα: 3,67 casos/100 pacientes‐ano vs. 2,16 casos/100 pacientes‐ano.47,48 Entretanto, esses dados ainda não estão disponíveis em doenças dermatológicas tratadas com iJAK.
Dados de vida real de pacientes com DA comparando os perfis de segurança do baricitinibe e tofacitinibe confirmaram HZ como evento adverso mais frequente (5,6% tofacitinibe×4,9% baricitinibe).40 O risco é maior em idosos e na coadministração com corticosteroides ou metotrexato.44 No entanto, são poucos os casos de herpes disseminado ou acometendo vários dermátomos, sem evidência de doença visceral ou óbito pela infecção herpética. Os estudos clínicos demonstraram que o upadacitinibe apresentou maior risco de HZ comparado a indivíduos que utilizavam imunobiológicos e DMARDS.44 Tal como nos iJAK de primeira geração, o upadacitinibe em pacientes com AR apresentou risco maior de HZ quando prescrito em doses elevadas, nas quais o HZ é mais provável ocorrer e ser mais grave e de natureza a envolver vários dermátomos.49
Assim, a vacinação contra o vírus do HZ é um meio importante, embora imperfeito, de reduzir o impacto da infecção pelo HZV.44 Porém, tanto a vacina contra varicela (Varivax®, para indivíduos não imunes com menos de 50 anos) quanto a vacina contra zóster (Zostavax® para indivíduos ≥ 50 anos) utilizam vírus vivos (assim como a da febre amarela) e, como tal, recomenda‐se administrar pelo menos três a quatro semanas antes do início dos iJAK.44
No Brasil, já está disponível a vacina recombinante contra o HZV, Shingrix®, para pacientes com mais de 50 anos, adultos maiores de 18 anos com imunossupressão ou que vão receber imunossupressores, como os iJAK. Essa vacina confere taxas de proteção de 91,3% para pessoas com 70 anos ou mais, e de 67,3% em imunocomprometidos com mais de 50 anos. A vacina é composta pelo antígeno glicoproteína E (gE) da superfície viral e deve ser oferecida em duas doses (0‐2 meses). Como não utiliza partículas vivas, pode ser administrada em concomitância com o tratamento iJAK.50
Estudos de eficácia sobre o uso de Shingrix® em uma população imunocomprometida ≥ 65 anos e em indivíduos tratados para DII com imunossupressores e ≥ 50 anos mostraram boa proteção contra HZ. Como ambos (iJAK e Shingrix®) estão recentemente disponíveis, o seguimento de longo prazo dessa associação deve revelar detalhes do perfil de imunização nesses pacientes.
Os iJAK reduzem as diferentes citocinas e a sinalização dos receptores do fator de crescimento, dependendo do tipo de inibição de JAK. Tofacitinibe, um inibidor de JAK1/3, tem efeitos importantes no desenvolvimento dos linfócitos B naïve. Isso sugere perda na capacidade de imunização contra novos antígenos. Encontrou‐se resposta imune diminuída à vacina pneumocócica polissacarídica 23‐valente (PPSV‐23), especialmente em pacientes recebendo metotrexato concomitantemente: tofacitinibe, 45,1% vs. controles saudáveis 68,4%. A interrupção temporária do tofacitinibe por duas semanas (uma semana antes e uma após a vacinação) não restaurou a eficácia da vacina pneumocócica. Já os resultados para a vacina da influenza não foram afetados pelo uso do tofacitinibe.51
No entanto, a maioria dos pacientes com psoríase tratada com tofacitinibe desenvolveu imunidade adequada contra infecção pneumocócica (vacina pneumocócica conjugada‐13) e vacina contra o tétano. A vacinação com a pneumocócica conjugada (PCV‐13) foi bem‐sucedida em 68% dos pacientes com AR tratados com o baricitinibe, enquanto apenas 43% alcançaram aumento ≥ 4×nas concentrações da IgG antitetânica. Contudo, a maioria dos pacientes também fazia uso de metotrexato (89%) e/ou corticosteroides (30%). Resultados semelhantes foram encontrados para upadacitinibe, com resposta humoral satisfatória (aumento ≥ 2×nos níveis de anticorpos) para a vacina PCV‐13 em 12 semanas em 65% e 55%,respectivamente, entre os pacientes em uso de 15mg/dia e 30mg/dia.51
Assim, a opinião dos autores desta revisão é de que todo o calendário vacinal seja atualizado ao menos três a quatro semanas antes do início do uso dos iJAK sistêmicos, conforme orientações da autoridade de saúde local, inclusive aquelas do Ministério da Saúde do Brasil. E, durante o tratamento com iJAK, seja cumprido o calendário vacinal (exceto para vírus vivos), sem interrupção do tratamento.
Tal como ocorre com outros imunossupressores e com imunobiológicos anti‐TNFα, há preocupação de infecção por Mycobacterium tuberculosis com o uso de iJAK,17 e todos os pacientes que vão iniciar seu uso devem ser examinados e investigados quanto à tuberculose latente ou ativa.44 O risco de infecção por tuberculose é dependente do risco epidemiológico de cada região. A incidência de tuberculose com tofacitinibe foi maior em regiões endêmicas (0,75/100 pacientes‐ano) em relação às regiões de menor risco (0,02/100 pacientes‐ano).44,52
Como o Brasil apresenta taxas endêmicas de tuberculose, que pode permanecer latente por décadas, a vigilância deve ocorrer previamente e durante o uso de imunossupressores como iJAK por meio do teste PPD (prova da tuberculina/Mantoux) ou dos ensaios de liberação da IFN (IGRA: ELISPOT, Quantiferon gold, Quantiferon gold plus), que apresentam performance discretamente superior.53,54 Testes de rastreio de tuberculose positivos devem ser referenciados ao infectologista a fim de considerar o tratamento da tuberculose latente com isoniazida, por nove meses, já que 5%‐10% apresentam ativação, especialmente em pacientes sob imunossupressão.
No âmbito do uso de iJAK, essas medicações conferem maior risco de infecções gerais, risco de infecções oportunistas como pneumonia por Pneumocystis jirovecii, HZ, tuberculose, citomegalovirose e infecções por Epstein‐Barr vírus. Recomenda‐se: 1) rastreamento de infecção pelo vírus da hepatite B previamente ao tratamento; 2) profilaxia para P. jirovecii em pacientes com fatores de risco adicionais, como uso de corticosteroides; 3) rastreamento de tuberculose latente; 4) monitorização da carga viral em pacientes HBsAg negativos porém anti‐HBcAg positivos em virtude do risco de reativação de infecção oculta pelo vírus da hepatite C; 5) ao longo do uso crônico de iJAK, os médicos devem estar atentos para o aumento de outras infecções oportunistas, como infecções fúngicas invasivas, especialmente em pacientes que já apresentam risco adicional por fatores como uso prévio ou concomitante de corticosteroides, baixa contagem de linfócitos no hemograma ou terapia dependente de altas doses de iJAK.55
Infecções fúngicas invasivas foram detectadas em EELP e nos ensaios com tofacitinibe em 15 casos entre 9.291 pacientes; infecção por Candida spp. foi a mais comum, seguida por critptococose, histplasmose e P. jirovecii.44
Eventos tromboembólicosBaricitinibe, tofacitinibe, ruxolitinibe e upadacitinibe incluem advertências para potencial eventos de trombose venosa profunda, embolia pulmonar e trombose arterial.56 Embora esses riscos pareçam ser baixos e dependentes da dose, estudos adicionais são necessários para determinar o mecanismo exato por trás de seus efeitos trombóticos.
A descoberta pelo estudo ORALSURV de risco aumentado de tromboembolismo venoso (TEV) com dose de 10mg/dia do tofacitinibe em relação aos anti‐TNFα corrobora esse fato, observado pela primeira vez em ensaios clínicos com pacientes com AR na dose de 4mg/dia de baricitinibe, o que fortalece o conceito de que o TEV pode ser um verdadeiro evento adverso relacionado ao uso de iJAK em pacientes com AR.44
Embora ainda falte plausibilidade biológica, é razoável especular que maior modulação de JAK2, observada com doses mais altas de tofacitinibe e baricitinibe, mediaria os efeitos trombóticos.44 Contudo, de modo tranquilizador, os iJAK usados na AR, em suas doses atualmente aprovadas, ainda não parecem apresentar risco excessivo de eventos tromboembólicos.44
O estudo ORALSURV relatou que 5mg/dia de tofacitinibe e o uso de anti‐TNFα estão associados a risco semelhante de TEV, e isso é consistente com dados do mundo real.44 Além disso, as taxas de incidência de TEV observadas nos estudos principais de tofacitinibe e upadacitinibe foram semelhantes (e foram menores nos estudos de filgotinibe), e as taxas nos grupos de comparadores ativos desses programas (metotrexato ou adalimumabe) não foram superiores. Mesmo para o baricitinibe, para o qual o desequilíbrio inicial no risco de TEV entre as doses de 2mg/dia e 4mg/dia nas primeiras 12 semanas dos ensaios de fase III levantou suspeitas, taxas de incidência de longo prazo semelhantes foram relatadas para ambas as doses, de 0,5/100 pacientes‐ano, em linha com estudos de base populacional de AR.44,57
Por fim, baricitinibe 4mg/dia administrado por duas semanas não aumentou o risco de TEV no tratamento da COVID‐19, uma condição per se com risco aumentado de TEV no início do estudo.58 Como as descobertas do ORALSURV sugerem haver risco dose‐dependente com tofacitinibe em relação aos anti‐TNFα, até que pesquisas mais fundamentadas sejam realizadas com cada um desses compostos, é prudente evitar os iJAK em pacientes com risco elevado de TEV, particularmente aqueles com história de TEV que não estejam atualmente anticoagulados.44
Em 2021, a Food and Drug Administration (FDA) emitiu uma comunicação de segurança e exigiu revisões de preucaução na embalagem para os iJAK tofacitinibe, baricitinibe e upadacitinibe, que incluía informações sobre o risco de eventos cardíacos graves, câncer, trombose e óbito.55
Neoplasias malignasA taxa geral de ocorrência de neoplasias malignas com o uso dos iJAK em ensaios clínicos randomizados envolvendo pacientes com AR e estudos de extensão de longo prazo foi semelhante à observada com bDMARD.33
No estudo ORALSURV, a taxa de incidência de malignidade (por 100 pacientes‐ano) foi de 1,13 para pacientes tratados com tofacitinibe 5mg, 2×/dia e 1,13 com tofacitinibe 10mg, 2×/dia, em comparação com 0,77 para os anti‐TNFα.33,44
O maior risco de malignidades foi impulsionado por taxas diferenciais de vários tipos de câncer (particularmente câncer de pulmão e linfoma), observados principalmente nos grupos norte‐americanos do estudo (em comparação com o restante do mundo), entre indivíduos mais idosos e com histórico de tabagismo.33 Também foi notado risco aumentado de câncer de pele não melanoma, observado anteriormente com o uso da dose de 10mg/dia de tofacitinibe na colite ulcerativa.59 Entretanto, taxas mais altas de melanoma foram observadas em pacientes usando anti‐TNFα 0,09 vs. 0,02 para qualquer dose do tofacitinibe.59
O mecanismo pelo qual os iJAK podem estar associados a alguns tipos de câncer é desconhecido, mas especula‐se que alguns iJAK, dependendo de sua seletividade e efeito nas células natural killer (NK), poderiam diminuir a imunovigilância do hospedeiro, tornando um câncer existente ou de novo com maior possibilidade de evolução, como ocorre com os outros imunossupressores60
Em resumo, quando os iJAK orais são avaliados de modo geral, as taxas de TEV relatadas variaram de nenhum evento a 0,1%‐0,5% em ensaios clínicos específicos de dermatologia de fase III em comparação com nenhum evento no placebo. As taxas de eventos cardiovasculares variaram de nenhum evento a 0,4%‐1,2% em comparação com nenhum evento a 0,5%‐1,2% no placebo. As taxas de infecções graves foram de 0,4%‐4,8% em comparação com nenhum evento a 0,5%‐1,3% no placebo. As taxas de câncer de pele não melanoma variaram de nenhum evento a 0,6%‐0,9% em comparação com nenhum evento no placebo. As taxas de câncer de pele não melanoma variaram de nenhum evento a 0,2%‐0,7% em comparação com nenhum evento a 0,6% no placebo. A maioria dos pacientes que desenvolveram esses eventos adversos tinha fatores de risco para o evento específico. Os EA mais habituais foram infecções das vias aéreas superiores, náuseas, cefaleia, acne e dislipidemia.61
Contraindicações aos iJAKA principal contraindicação aos iJAK é a incerteza no diagnóstico da doença inflamatória que se deseja tratar.62 Várias dermatoses inflamatórias podem simular linfoma cutâneo de células T, como DA e psoríase; alopecias podem se relacionar a sífilis; hidradenite supurativa, a processos infecciosos como tuberculose, actinomicose, paracoccidioidomicose e doença de Crohn metastática. O uso inadvertido dos iJAK nessas condições pode desencadear desfechos graves.
Outras contraindicações dos iJAK incluem: (i) hipersensibilidade aos componentes das medicações (p. ex., Tween 80); (ii) tuberculose; (iii) função renal alterada (baricitinibe); (iv) alteração grave da função hepática (upadacitinibe e abrocitinibe); (v) neutropenia <500 células/mm3 (baricitinibe) ou <1.000 células/mm3 (upadacitinibe e abrocitinibe); (vi) anemia (hemoglobina ≤ 9g/dL); (vii) processos infecciosos ativos; (viii) gestantes, mulheres com desejo de engravidar ou com vida sexual ativa sem uso de método contraconceptivo; (ix) plaquetopenia <50.000mm3 (abrocitinibe).62
A prescrição também deverá ser reconsiderada caso o paciente se mostre impossibilitado de realizar exames de rastreamento infeccioso antes do início do tratamento ou acompanhamento regular durante o tratamento.
Avaliação dos pacientes antes e durante o uso dos iJAKAnamnese e exame clínico geral, além do dermatológico, devem ser realizados em virtude das potenciais complicações com doenças infecciosas virais, bacterianas e fúngicas, além de neoplasias.
Avaliar histórico de perda ponderal de causa não esclarecida, contatos profissionais ou familiares com doenças infectocontagiosas (tuberculose, doença de Chagas, hanseníase), não realização de exames periódicos de prevenção de câncer indicados para a faixa etária do paciente e antecedentes familiares de câncer ou de erros inatos da imunidade (imunodeficiências).62
A palpação das cadeias linfonodais, fígado, baço, ausculta pulmonar e cardíaca, sinal de Giordano são essenciais a cada consulta. Outros dados a serem verificados antes do tratamento estão incluídos na tabela 5.
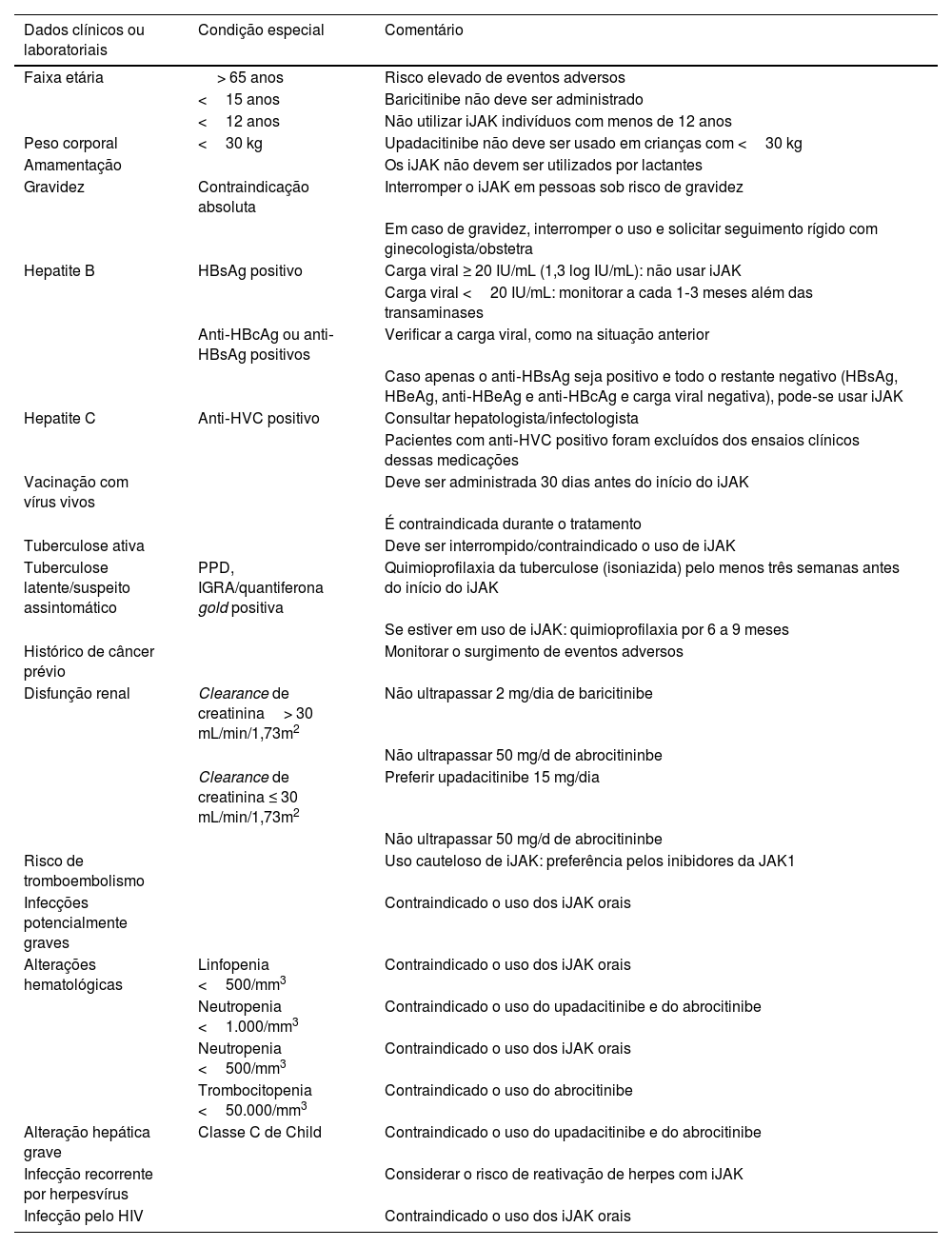
Considerações clínicas e laboratoriais sobre o uso dos inibidores da via JAK‐STAT
| Dados clínicos ou laboratoriais | Condição especial | Comentário |
|---|---|---|
| Faixa etária | > 65 anos | Risco elevado de eventos adversos |
| <15 anos | Baricitinibe não deve ser administrado | |
| <12 anos | Não utilizar iJAK indivíduos com menos de 12 anos | |
| Peso corporal | <30 kg | Upadacitinibe não deve ser usado em crianças com <30 kg |
| Amamentação | Os iJAK não devem ser utilizados por lactantes | |
| Gravidez | Contraindicação absoluta | Interromper o iJAK em pessoas sob risco de gravidez |
| Em caso de gravidez, interromper o uso e solicitar seguimento rígido com ginecologista/obstetra | ||
| Hepatite B | HBsAg positivo | Carga viral ≥ 20 IU/mL (1,3 log IU/mL): não usar iJAK |
| Carga viral <20 IU/mL: monitorar a cada 1‐3 meses além das transaminases | ||
| Anti‐HBcAg ou anti‐HBsAg positivos | Verificar a carga viral, como na situação anterior | |
| Caso apenas o anti‐HBsAg seja positivo e todo o restante negativo (HBsAg, HBeAg, anti‐HBeAg e anti‐HBcAg e carga viral negativa), pode‐se usar iJAK | ||
| Hepatite C | Anti‐HVC positivo | Consultar hepatologista/infectologista |
| Pacientes com anti‐HVC positivo foram excluídos dos ensaios clínicos dessas medicações | ||
| Vacinação com vírus vivos | Deve ser administrada 30 dias antes do início do iJAK | |
| É contraindicada durante o tratamento | ||
| Tuberculose ativa | Deve ser interrompido/contraindicado o uso de iJAK | |
| Tuberculose latente/suspeito assintomático | PPD, IGRA/quantiferona gold positiva | Quimioprofilaxia da tuberculose (isoniazida) pelo menos três semanas antes do início do iJAK |
| Se estiver em uso de iJAK: quimioprofilaxia por 6 a 9 meses | ||
| Histórico de câncer prévio | Monitorar o surgimento de eventos adversos | |
| Disfunção renal | Clearance de creatinina> 30 mL/min/1,73m2 | Não ultrapassar 2 mg/dia de baricitinibe |
| Não ultrapassar 50 mg/d de abrocitininbe | ||
| Clearance de creatinina ≤ 30 mL/min/1,73m2 | Preferir upadacitinibe 15 mg/dia | |
| Não ultrapassar 50 mg/d de abrocitininbe | ||
| Risco de tromboembolismo | Uso cauteloso de iJAK: preferência pelos inibidores da JAK1 | |
| Infecções potencialmente graves | Contraindicado o uso dos iJAK orais | |
| Alterações hematológicas | Linfopenia <500/mm3 | Contraindicado o uso dos iJAK orais |
| Neutropenia <1.000/mm3 | Contraindicado o uso do upadacitinibe e do abrocitinibe | |
| Neutropenia <500/mm3 | Contraindicado o uso dos iJAK orais | |
| Trombocitopenia <50.000/mm3 | Contraindicado o uso do abrocitinibe | |
| Alteração hepática grave | Classe C de Child | Contraindicado o uso do upadacitinibe e do abrocitinibe |
| Infecção recorrente por herpesvírus | Considerar o risco de reativação de herpes com iJAK | |
| Infecção pelo HIV | Contraindicado o uso dos iJAK orais |
iJAK, inibidor da via JAK‐STAT; PPD, teste tubercuínico intradérmico; IGRA, teste tuberculínico de liberação de interferona.
Antes do tratamento e periodicamente, a cada dois meses, os pacientes em uso de iJAK sistêmicos devem realizar exames de sangue (hemograma, lipidograma, função hepática e renal), radiografia do tórax antes do tratamento, repetindo‐se quando indicado.
Manejo de eventos durante o tratamento com os iJAKOs iJAK podem promover aumento dos lípides séricos, com aumento do colesterol total, LDL, HDL e triglicérides.62 Os níveis do colesterol e triglicérides devem ser monitorados durante o tratamento, de modo que, quando necessário, antilipemiantes orais sejam utilizados, de acordo com os protocolos clínicos.63
É comum a elevação da creatinofosfoquinase (CPK) durante o tratamento, geralmente não relacionado à miopatia, o que demanda atenção quanto aos resultados no seguimento laboratorial.62 Postula‐se que o aumento nos níveis da CPK durante o uso do updacitinibe ocorra porque o iJAK1 inibe a ação fisiológica da oncostatina M (OSM), que bloqueia a diferenciação dos mioblastos a miócitos, onde se produz mais CPK. Uma vez que se perca essa via inibidora, mais mioblastos se diferenciam em miócitos e aumenta a síntese da CPK, sem rabdomiólise, apenas pela diferenciação celular. Acredita‐se que inclusive a OSM em níveis elevados determine a sarcopenia nos pacientes com AR. A suspeita de rabdomiólise decorrente do iJAK deve seguir mialgia, câimbras e escurecimento da urina, configurando urgência clínica.62
Pacientes recebendo iJAK devem ser monitorados quanto à elevação das transaminases e orientados a procurar atendimento médico caso apresentem sintomas sistêmicos: náusea/vômitos/anorexia, como sintomas digestivos, e erupção cutânea/prurido/icterícia.62
O risco de citopenias é maior entre pacientes> 65 anos. O iJAK deve ser interrompido em casos de neutropenia (< 1.000 neutrófilos/mm3), linfopenia (< 500 linfócitos/mm3), anemia (hemoglobina <8g/dL) ou plaquetopenia (< 50.000mm3).62 Neutropenia e linfopenia aumentam os riscos de infecção, e o paciente deve ser orientado a procurar atendimento médico em caso de febre, calafrios ou dor de garganta ou tosse. Caso ocorram sintomas de fadiga, mal‐estar, palpitações, tonturas ou dispneia, dor abdominal de início abrupto, náuseas, vômitos ou anorexia, o paciente também deve buscar atendimento médico, pelo risco de perfuração intestinal. Em pacientes com doença diverticular do cólon, a perfuração gastrintestinal pode suceder uma diverticulite; esses pacientes devem ser instruídos e cautelosamente acompanhados.62
Sintomas como tosse seca, dispneia ao exercício e febre devem alertar o paciente a procurar atendimento médico, sob risco de pneumopatia intersticial. Nesse caso, exames de sangue devem ser solicitados: PCR, DHL, Krebs von den Lungen‐6 (KL‐6) e SP‐D (surfactante protein D) – os níveis séricos dessas duas últimas são muito sensíveis no diagnóstico de pneumonite de hipersensibilidade medicamentosa.64 Além disso, radiografia ou tomografia computadorizada do tórax e gasometria devem ser solicitados, devendo ser descontinuado o iJAK até o diagnóstico.
Pneumonia por P. jirovecci pode ocorrer, e a dosagem dos níveis de β‐D‐glucana podem auxiliar o diagnóstico. Além disso, deve‐se afastar influenza, COVID‐19, infecções por Mycoplasma, Chlamydia e Legionella em pacientes com imagens radiológicas de padrão parenquimatoso intersticial, o que pode configurar pneumonia atípica não pneumocística. Excluídas causas infecciosas, pneumonia de doença pulmonar reumática induzida por medicamento deve ser considerada. Imagens radiológicas exsudativas no pulmão podem indicar pneumonia bacteriana ou tuberculose.62
iJAK nas dermatoses inflamatórias e autoimunesA maior parte das dermatoses inflamatórias ou autoimunes não decorre do efeito de uma única citocina, porém, segue perfis inflamatórios que podem ser inibidos pela modulação da via JAK‐STAT. A base imunológica e os resultados clínicos dos principais iJAK, na dermatologia, são apresentadas adiante. A tabela 6 apresenta os iJAK com indicações dermatológicas descritas na literatura, e a figura 2 resume as principais substâncias com ensaios clínicos para uso na dermatologia.
Indicações dermatológicas para os inibidores da via JAK‐STAT, relatadas na literatura
| iJAK | DA | PSO | VITI | AA | LP | LE | DM | VL | PG | GA | NL | SARC | MOR | HS | DEVH | ECZm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oral | ||||||||||||||||
| Tofacitinibe | X | X | X | X | X | X | X | X | X | X | X | |||||
| Baricitinibe | X | X | X | X | X | |||||||||||
| Abrocitinibe | X | |||||||||||||||
| Upadacitinibe | X | X | ||||||||||||||
| Ruxolitinibe | X | X | X | X | X | |||||||||||
| Delgocitinibe | X | |||||||||||||||
| Ritlecitinibe | X | X | ||||||||||||||
| Elsubritinibe | X | |||||||||||||||
| Itacitinibe | X | X | ||||||||||||||
| Peficitinibe | X | |||||||||||||||
| Deucravacitinibe | X | |||||||||||||||
| Filotinibe | X | |||||||||||||||
| Solcitinibe | X | |||||||||||||||
| Pacritinibe | X | |||||||||||||||
| Deuruxolitinibe | X | |||||||||||||||
| Gusacitinibe | X | |||||||||||||||
| Tópico | ||||||||||||||||
| Tofacitinibe | X | X | X | X | X | |||||||||||
| Ruxolitinibe | X | X | X | X | X | |||||||||||
| Brepocitinibe | X | |||||||||||||||
| Delgocitinibe | X | X | ||||||||||||||
| Cerdulatinibe | X |
iJAK, inibidor da via JAK‐STAT; AA, alopecia areata; DA, dermatite atópica; DM, dermatomiosite; PSO, psoríase; VITI, vitiligo; HS, hidradenite supurativa; LP, líquen plano e líquen plano pilar; LE, lúpus eritematoso; PG, pioderma gangrenoso; GA, granuloma anular; SARC, sarcoidose; NL, necrobiose lipoídica; VL, vasculopatia livedoide; MOR, morfeia; ECZm, eczema crônico das mãos; DEVH, doença enxerto versus hospedeiro.

Diagrama representando os principais iJAK que apresentam resultados favoráveis em estudos clínicos para dermatoses inflamatórias e autoimunes. AA, alopecia areata; DA, dermatite atópica; DM, dermatomiosite; PSO, psoríase; VITI, vitiligo; HS, hidradenite supurativa; DEVH, doença enxerto versus hospedeiro; Abro, abrocitinibe; Upa, upadacitinibe; Bari, baricitinibe; DelgoT, delgocitinibe tópico; Tofa, tofacitinibe; RuxoT, ruxolitinibe tópico; Ruxo, ruxolitinibe; Ritle, ritlecitinibe; Deucra, deucravacitinibe; Solci, solcitinibe; Itaci, itacitinibe; Deuruxo, deuruxolitinibe
A DA apresenta alta prevalência, especialmente em crianças e adultos jovens. Seu curso crônico e redicivante, associado a comorbidades alérgicas e psicológicas, infligem importante impacto na qualidade de vida.65 Sua patogênese é complexa e depende da interação de vários elementos. Em cerca de 80% dos pacientes ocorrem falha da barreira cutânea, especialmente por perda de função do gene da filagrina. Perda transepidérmica de água, alterações no microbioma cutâneo, em conjunto com permeabilidade a antígenos e hiper‐reatividade neural favorecem a exacerbação da resposta inflamatória local, manifesta especialmente como eczema e prurido.66,67
Apesar de ser considerada doença inflamatória com predominância do padrão Th2, diversas citocinas atuam conjuntamente para formar e manter um ambiente de hiper‐reatividade cutânea e que variam também entre as fases da doença.3 O envolvimento no microambiente cutâneo, especialmente de IL‐4, IL‐5, IL‐10, IL‐13, IL‐31, IFN‐γ, IL‐17, IL‐22 e IL‐33, é indício do potencial dos iJAK, (principalmente JAK1) em seu tratamento.67,68
Modelos experimentais demonstraram que o tratamento da DA com iJAK levou à rápida supressão do prurido, melhora da barreira cutânea, das fibras nervosas cutâneas, redução do infiltrado linfocitário e das citocinas inflamatórias (p. ex., IL‐4 e IL‐13).69
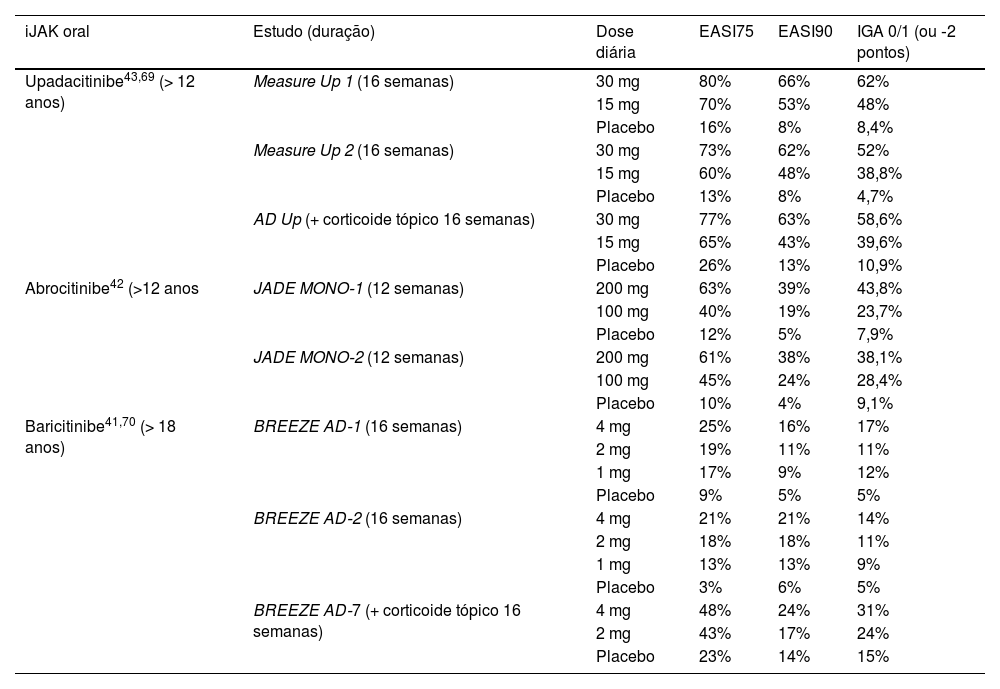
No Brasil, baricitinibe, abrocitinibe e upadacitinibe são aprovados para uso oral na DA moderada a grave; ruxolitinibe 1,5% creme está disponível no mercado dos EUA, e delgocitinibe 0,5% creme deve ser aprovado em 2023. Apesar de eficaz no tratamento da DA, não foram desenvolvidos ensaios clínicos com tofacitinibe oral – a maioria dos estudos é de relatos e séries de casos.67 A tabela 7 resume os resultados de ensaios clínicos pivotais dos principais iJAK na DA.
Resultados dos principais estudos clínicos sobre os inibidores da via JAK‐STAT utilizados no tratamento da dermatite atópica
| iJAK oral | Estudo (duração) | Dose diária | EASI75 | EASI90 | IGA 0/1 (ou ‐2 pontos) |
|---|---|---|---|---|---|
| Upadacitinibe43,69 (> 12 anos) | Measure Up 1 (16 semanas) | 30 mg | 80% | 66% | 62% |
| 15 mg | 70% | 53% | 48% | ||
| Placebo | 16% | 8% | 8,4% | ||
| Measure Up 2 (16 semanas) | 30 mg | 73% | 62% | 52% | |
| 15 mg | 60% | 48% | 38,8% | ||
| Placebo | 13% | 8% | 4,7% | ||
| AD Up (+ corticoide tópico 16 semanas) | 30 mg | 77% | 63% | 58,6% | |
| 15 mg | 65% | 43% | 39,6% | ||
| Placebo | 26% | 13% | 10,9% | ||
| Abrocitinibe42 (>12 anos | JADE MONO‐1 (12 semanas) | 200 mg | 63% | 39% | 43,8% |
| 100 mg | 40% | 19% | 23,7% | ||
| Placebo | 12% | 5% | 7,9% | ||
| JADE MONO‐2 (12 semanas) | 200 mg | 61% | 38% | 38,1% | |
| 100 mg | 45% | 24% | 28,4% | ||
| Placebo | 10% | 4% | 9,1% | ||
| Baricitinibe41,70 (> 18 anos) | BREEZE AD‐1 (16 semanas) | 4 mg | 25% | 16% | 17% |
| 2 mg | 19% | 11% | 11% | ||
| 1 mg | 17% | 9% | 12% | ||
| Placebo | 9% | 5% | 5% | ||
| BREEZE AD‐2 (16 semanas) | 4 mg | 21% | 21% | 14% | |
| 2 mg | 18% | 18% | 11% | ||
| 1 mg | 13% | 13% | 9% | ||
| Placebo | 3% | 6% | 5% | ||
| BREEZE AD‐7 (+ corticoide tópico 16 semanas) | 4 mg | 48% | 24% | 31% | |
| 2 mg | 43% | 17% | 24% | ||
| Placebo | 23% | 14% | 15% |
| iJAK tópico | Estudo (duração) | Concentração 2×/dia | EASI 75 | EASI 90 | IGA 0/1 (ou–2 pontos) |
|---|---|---|---|---|---|
| Ruxolitinibe71 | TRuE ‐ AD 1 (8 semanas) | 1,5% creme | 62,1% | 53,8% | |
| 0,75% creme | 56,0% | 50,0% | |||
| Veículo | 24,6% | 15,1% | |||
| TRuE ‐ AD 2 (8 semanas) | 1,5% creme | 61,8% | 51,3% | ||
| 0,75% creme | 51,5% | 39,0% | |||
| Veículo | 14,4% | 7,6% | |||
| Tofacitinibe72 | NCT02001181 (4 semanas) | 2% pomada | 68,0% | ||
| Veículo | 13,0% | ||||
| Delgocitinibe73,74 | OBA 4‐1 (4 semanas) | 0,5% creme | 26,4% | Corpo – 10,4% | |
| Face – 22,8% | |||||
| Veículo | 5,8% | Corpo – 3,8% | |||
| Face – 4,0% | |||||
| JapicCTI‐184064 (4 semanas) | 0,5% creme | 37,7% | |||
| Veículo | 4,4% |
EASI75 e EASI90, redução de ≥ 75%/ ≥ 90% no índice de área e gravidade do eczema; IGA 0/1, Avaliação Global do Investigador resulta em mínimo eritema e pápulas perceptíveis.
Nos estudos clínicos, todos os iJAK sistêmicos tiveram desempenho superior ao placebo, com rápido controle do prurido, melhora da qualidade de vida e do sono e redução da área de eczema. Doses mais altas e associação dos iJAK com corticosteroides tópicos promoveram melhor desempenho dos tratamentos, porém maior taxa de EA.
Na comparação dos estudos individuais, abrocitinibe e upadacitinibe orais apresentaram maior proporção de redução de escores clínicos que baricitinibe.40,76 Apesar das diferenças entre os estudos nas faixas etárias e etnias dos pacientes, na duração e nas diferentes estratégias de resgate, espera‐se que ≥ 60% daqueles em uso de upadacitinibe nas doses de 15mg/dia e ≥ 70% na dose de 30mg/dia atinjam a redução ≥ 75% do Índice de Área e Gravidade do Eczema (EASI75).40,43 Já no abrocitinibe 100mg/dia e 200mg/dia, espera‐se que esses valores sejam ≥ 40% e ≥ 60%. E, no baricitinibe 2mg/dia e 4mg/dia, que sejam ≥ 18% e ≥ 21%.40
A extensão por 52 semanas do estudo AD UP incluiu 901 pacientes entre 12 e 75 anos, com DA moderada ou grave, randomizados em três grupos: upadacitinibe 15mg/dia, 30mg/dia e placebo (primeiras 16 semanas), todos associados a corticosteroide tópico de baixa potência ou inibidor de calcineurina, 2×/dia. Na semana 52, a proporção de pacientes que atingiu EASI75 foi de 50,8% para o grupo upadacitinibe 15mg/dia e 69,0% para o grupo de 30mg/dia; 7,4% dos pacientes do grupo upadacitinibe 15mg/dia e 2,5% do grupo de 30mg/dia abandonaram o estudo por falta de eficácia.70
No estudo BREEZE‐AD3, que faz o seguimento por 52 semanas de 292 pacientes do estudo BREEZE‐AD7 (16 semanas) em uso de baricitinibe 2 ou 4mg/dia e corticosteroides tópicos, observou‐se uma leve perda de eficácia: EASI75 51% na semana 16 para 43% na semana 68. Até 17,8% dos pacientes tratados com baricitinibe 2mg/dia e 28,4% dos tratados com baricitinibe 4mg/dia abandonaram o seguimento por falta de eficácia.77
Em estudo de vida real que incluiu 44 pacientes adultos com DA tratados com baricitinibe, confirmou‐se a eficácia e perfil de segurança do medicamento; entretanto, o seguimento longitudinal revelou que o percentual de pacientes que atingiu EASI75 foi de 53% na semana 4, e 33% na semana 16; 7% dos pacientes abandonaram o estudo por eficácia insuficiente.78 Esses dados apontam para a possibilidade de uma leve perda de eficácia dos tratamentos a partir da quarta semana.
Ainda não há estudos randomizados comparando os iJAK orais a outros imunossupressores (p. ex., ciclosporina, azatioprina e metotrexato) na DA. Também não são disponíveis dados relativos à associação de iJAK tópico e sistêmico ou esquemas posológicos variáveis, como a utilização de doses altas nas primeiras semanas, com posterior redução das doses ou troca de estratégia para minimizar custo e EA.
O estudo Heads up comparou 692 adultos com DA, randomizados para usar upadacitinibe oral 30mg/dia vs. dupilumabe 300mg a cada 14 dias, por 24 semanas. Na semana 16, o EASI75 foi atingido por 71% dos pacientes usando upadacitinibe e por 61% daqueles em uso de dupilumabe. Os demais desfechos clínicos seguiram essa superioridade do upadacitinibe, com destaque à maior e mais precoce redução do prurido. Na semana 24, o EASI75 do grupo upadacitinibe reduziu para 64%, enquanto o dupilumabe resultou em 59%. Ambos os grupos demandaram tratamentos de resgate antes da semana 16: upadacitinibe 20,3% e dupilumabe 17,5%.79
Nessas doses, ao fim de 24 semanas, o estudo evidenciou maior frequência de acne (18,4% vs. 3,2%), HSV (0,9% vs. 0,0%) e HZ (3,4% vs. 1,2%) no grupo upadacitinibe, enquanto conjuntivite foi mais frequente no grupo dupilumabe (10,2% vs. 1,4%). Um participante do grupo upadacitinibe faleceu em decorrência de broncopneumonia associada à infecção pelo vírus influenza. No grupo upadacitinibe, houve maior frequência de alterações laboratoriais. Elevação das transaminases ocorreu em 3,4% vs. 1,1%, e dois participantes do grupo upadacitinibe descontinuaram o estudo por esse motivo. Anemia (2,0% vs. 0,3%), neutropenia (1,7% vs. 0,6%) e elevação da CPK (7,5% vs. 3,2%) também prevaleceram no upadacitinibe.79 O perfil de segurança de longo prazo do dupilumabe permite recomendar sua administração sem a necessidade de seguimento laboratorial ou risco de interação medicamentosa.67,76
O estudo JADE DARE comparou abrocitinibe 200mg/dia vs. dupilumabe 300mg a cada 14 dias, em 726 pacientes com DA moderada a grave, por 26 semanas. Ao final do estudo, 73% dos pacientes do grupo abrocitinibe vs. 72% do grupo dupilumabe atingiram o EASI75. Conjuntivite foi mais prevalente no grupo dupilumabe (11% vs. 3%). No grupo abrocitinibe, foram mais frequentes: acne (13% vs. 3%), náuseas (19% vs. 2%), HSV (6% vs. 5%), HZ (3% vs. 0%) e cefaleia (13% vs. 7%).80
Os iJAK tópicos (ruxolitinibe e delgocitinibe) ratificaram o rápido início de efeito (< 24 horas para redução do prurido), com alta taxa de eficácia, segurança e tolerabilidade. Até o momento, iJAK tópicos são indicados em quadros com menos de 20% da área corporal afetada e uso não prolongado.18,75,81 A eficácia dos iJAK tópicos aumenta com a concentração do ativo e o número de aplicações diárias. A boa tolerabilidade e o perfil de segurança podem revelar‐se oportunos no resgate de pacientes jovens com DA moderada a grave, que geralmente dependem de corticosteroides tópicos para tal. O uso estendido de ruxolitinibe 1,5% creme por até 44 semanas, com 1.249 adultos orientados a usar nas lesões, se necessário, resultou em bom perfil de tolerabilidade e segurança, sem evidências de efeito sistêmico. Entre 74% e 78% atingiram o fim do estudo com IGA 0/1.82
Quando comparados os desempenhos dos iJAK tópicos em estudos clínicos individuais, tofacitinibe 2%, delgocitinibe 3% e ruxolitinibe 1,5% apresentaram maior redução de escores de DA que os esperados para os inibidores da PDE4 (p. ex., crisaborol).78,83
Em uma comparação duplo‐cega envolvendo 51 pacientes com DA usando triancinolona creme 0,15%, 2×/dia, sob diferentes concentrações e regimes de ruxolitinibe creme por oito semanas, tanto a triancinolona quanto as diferentes posologias de ruxolitinibe evidenciaram superioridade em relação ao veículo e incremento do efeito em função do tempo. EASI75 ocorreu em 56% dos pacientes do grupo que utilizou ruxolitinibe 1,5%, 2×/dia vs. 47% dos que usaram triancinolona.84
De modo geral, os iJAK ainda não são liberados para crianças menores de 12 anos, exatamente a população mais afligida pela DA, porém os estudos de segurança têm avançado para ampliar a faixa etária de uso.
Por fim, os iJAK orais ou tópicos revelaram‐se opções terapêuticas valiosas para a DA, com bom perfil de segurança, porém sua indicação em DA precisa ser bem definida frente aos demais imunossupressores, imunobiológicos e fototerapia.
PsoríaseA patogênese da psoríase envolve tanto o sistema imune inato quanto o adaptativo, a partir de uma complexa interação entre queratinócitos, células dendríticas, linfócitos T, citocinas e outros mediadores inflamatórios. A inflamação crônica característica da doença é, principalmente, promovida pela IL‐23, a qual induz a diferenciação de células T naïve em linfócitos Th17 e sua expansão clonal.85,86
A transdução do sinal da IL‐23 é mediada pela TYK2. As células Th17 ativadas liberam citocinas proinflamatórias como TNFα, IL‐22, IL‐26 e IL‐29, as quais induzem a proliferação dos queratinócitos. Na psoríase, há ainda níveis séricos aumentados de citocinas Th1 (IFN‐γ e IL‐2), bem como níveis diminuídos de IL‐10. A IL‐22, produzida pelas células Th22, é citocina mediada pela via JAK‐STAT e induz a proliferação queratinocítica. A liberação da IL‐22, em associação à IL‐15, é sinalizada pela JAK1 e JAK3. Desse modo, a inibição da JAK/TYK é alvo potencial para seu tratamento. Contudo, face à patogênese da doença, os iJAK de segunda geração, seletivos para JAK2/TYK2, seriam potencialmente mais efetivos do que os que bloqueiam múltiplas isoformas de JAK/TYK.1,87 A propósito, indivíduos com polimorfismo genético e consequente perda da função da TYK2 apresentam menor risco de desenvolver psoríase e outras doenças imunomediadas.88
De maneira geral, tofacitinibe (2, 5, 10 e 15mg, 2×/dia), solcitinibe (200 e 400mg, 2×/dia), baricitinibe (8 e 10mg, 2×/dia) e deucravacitinibe (3 e 6mg, 2×/dia) apresentaram resposta PASI75 superior ao placebo, tanto na semana 8 quanto na 12, nos ensaios clínicos randomizados de psoríase em placas moderada à grave.89,90
Uma metanálise concluiu que tofacitinibe (15 e 10mg, 2×/dia) e deucravacitinibe (6mg, 2×/dia e 12mg/dia) apresentaram as melhores respostas de PGA e PASI75 (nas semanas 8 e 12), dentre os iJAK utilizados no tratamento da psoríase em placas. Apesar de a mesma metanálise mostrar perfil satisfatório de segurança dos iJAK no tratamento da psoríase em placa, tofacitinibe não obteve aprovação pela FDA em virtude dos efeitos colaterais apresentados; o medicamento é aprovado apenas na dose de 5mg, 2×/dia, para a terapia da AR. Os iJAK em estudo para o tratamento da psoríase em placas são deucravacitinibe, brepocitinibe e ropsacitinibe, todos inibidores da TKY2.89
No Brasil, tofacitinibe 5mg, 2×/dia, é aprovado para o tratamento da artrite psoriásica em adultos. Contudo, pode ser eficaz no tratamento da psoríase ungueal; 33% dos pacientes atingiram NAPSI50 na semana 16 com tofacitinibe 5mg, 2×/dia, 44% com 10mg, 2×/dia, e 12% com placebo.91
Atualmente, o único iJAK tópico em estudo para o tratamento da psoríase é o brepocitinibe, inibidor ortostérico da JAK1 e JAK2. Em ensaio fase I envolvendo indivíduos sadios e doentes com psoríase em placas moderada a grave, o brepocitinibe confirmou segurança e boa tolerabilidade.92
Em comparação ao apremilaste, único outro medicamento oral aprovado para a terapia da psoríase, os inibidores da JAK2/TYK2 apresentam maior seletividade imunológica, restringindo a possibilidade de efeitos colaterais. O deucravacitinibe foi aprovado pela FDA e pela agência japonesa para o tratamento da psoríase em placa moderada a grave em adultos.93 Estudo randomizado com 332 pacientes usando 6mg/dia revelou resposta PASI75 superior, na semana 16, ao apremilaste oral 30mg, 2×/dia e placebo (58%, 35% e 13%). A taxa de efeitos colaterais foi similar nos três grupos. Entretanto, não se recomenda associar deucravacitinibe com outros imunossupressores.94
Alopecia areataA alopecia areata (AA) tem patogênese autoimune envolvendo o colapso do privilégio imunológico no folículo piloso. Nas áreas de AA, observa‐se infiltrado inflamatório composto por linfócitos CD4+, CD8+ e NK ao redor do bulbo dos folículos em fase anágena, levando à interrupção da melanogênese e da produção da haste.95 A presença dos linfócitos TCD8+NKG2D+ é determinante para o desenvolvimento das lesões de alopecia.96
IFN‐γ e IL‐15 induzem, principalmente, resposta citotóxica do tipo I. Linfócitos T CD8+ produzem IFN‐γ, que contribuem para a quebra do privilégio imunológico, induzindo a produção de IL‐15 pelas células epiteliais, que por sua vez leva à ativação das células efetoras CD8+NKGD2+ com mais produção de IFN‐γ fechando o ciclo que perpetua a doença. Tanto a ação da IFN‐γ na célula epitelial quanto da IL‐15 nas células CD8+NKG2D+ são mediadas pela via JAK‐STAT.96
A partir do relato de repilação de AA com tofacitinibe empregado no tratamento de psoríase,97 múltiplas séries de casos e estudos abertos não comparativos foram publicados mostrando resultados favoráveis com uso dos iJAK sistêmicos para tratamento da AA grave. O medicamento utilizado com mais frequência foi o tofacitinibe, seguido por ruxolitinibe e baricitinibe.98,99
Em estudo comparativo aberto, 75 pacientes com AA comprometendo mais de 30% do couro cabeludo foram randomizados em dois grupos: tofacitinibe 10mg/dia e ruxolitinibe 40mg/dia. Após seis meses de tratamento, ambos apresentaram melhora, sem diferença entre eles. O escore SALT50 foi atingido por 84% dos pacientes no grupo ruxolitinibe e 78% do grupo tofacitinibe, em seis meses.100
Em dois ensaios duplo‐cegos (BRAVE‐AA1, BRAVE‐AA2), pacientes com AA grave foram randomizados em três grupos: baricitinibe 2mg/dia, 4mg/dia ou placebo. Um total de 1.200 pacientes foi incluído para a análise de fase III desses estudos, na semana 36. O escore SALT20 foi atingido por 20% e 34% nos braços de 2 e 4mg/dia de baricitinibe, em comparação com 4% no placebo.101 A porcentagem de pacientes que descontinuou o tratamento por EA foi semelhante entre os grupos. Em 2022, o baricitinibe recebeu aprovação da FDA para o tratamento da AA.
No estudo ALLEGRO, 718 pacientes foram randomizados em seis grupos: ritlecitinibe 200mg/dia por quatro semanas e após 50mg/dia por 20 semanas; ritlecitinibe 200mg/dia por quatro semanas e após 30mg/dia por 20 semanas; ritlecitinibe 50mg/dia por 24 semanas; ritlecitinibe 10mg/dia por 24 semanas, ou placebo. A resposta ao ritlecitinibe foi dose‐dependente e 31% dos que usaram 50mg/dia após dose de ataque atingiram SALT20 na semana 24.102
Em um ensaio fase II, 149 pacientes foram divididos em quatro grupos: deuruxolitinibe 12, 8, 4mg/dia e placebo. Houve resposta dose‐dependente: 42%, 26%, 14% e 7% (placebo) dos pacientes atingiram SALT20 na semana 24.103
Um ensaio duplo‐cego recrutou 16 participantes com AA universal que foram orientados a aplicar tofacitinibe 2% pomada, ruxolitinibe 1% pomada, clobetasol 0,05% pomada e placebo, em diferentes locais do couro cabeludo. Seis pacientes (38%) em uso de tofacitinibe, cinco (31%) do ruxolitinibe e 10 (63%) do clobetasol tiveram repilação parcial nas áreas de tratamento, enquanto nenhum teve crescimento com placebo.104
Em estudo aberto, o ruxolitinibe 1,5% creme não se mostrou eficaz comparado ao placebo.105 Delgocitinibe 3% em pomada também não apresentou melhora no SALT após 12 semanas de tratamento.106 A baixa eficácia dos iJAK tópicos pode ser explicada pela dificuldade de penetração em camadas profundas da pele, não permitindo alcançar a inflamação peribulbar. Veículos que facilitem a penetração cutânea podem trazer perspectivas para os iJAK na AA.
O tratamento da AA grave é desafiador. Muitos pacientes não respondem à terapia convencional com imunossupressores sistêmicos ou são limitados pelos EA. Os casos graves apresentam altas taxas de recidiva após a suspensão do tratamento. Em consenso organizado pela Sociedade Brasileira de Dermatologia (2020), orienta‐se o uso dos iJAK sistêmicos como opção para AA extensa nos casos refratários à terapia convencional.107 Deve‐se destacar que, nos estudos que permitiram a aprovação do baricitinibe pela FDA, menos de 40% dos pacientes atingiram SALT20. Além disso, pacientes com mais de oito anos de doença (pior prognóstico) não foram incluídos. Estudos comparando iJAK sistêmicos com outros imunossupressores são essenciais para estabelecer reais benefícios frente às terapias disponíveis.
Na maioria dos casos, a manutenção dos resultados alcançados na AA depende da continuidade do tratamento. Estudos com maior período de seguimento são necessários para determinar a eficácia e segurança dos iJAK a longo prazo.
VitiligoO vitiligo afeta indivíduos suscetíveis geneticamente, e a autoimunidade é a principal causa da agressão aos melanócitos. As hipo/acromias, localizadas ou generalizadas, são as manifestações da destruição dos melanócitos dirigida pelos linfócitos T citotóxicos.108
As duas principais citocinas envolvidas na patogênese do vitiligo são a IFN‐γ e a IL‐15.109 A primeira é produzida pelos linfócitos T de memória (TRM) presentes na pele despigmentada e se liga ao receptor de IFN‐γ para estimular a expressão de quimiocinas como CXCL9, CXCL10 e CXCL11, que promovem a infiltração de células T autorreativas.110 A IL‐15 é produzida principalmente por queratinócitos, e está envolvida na sinalização para geração e manutenção das TRM na pele lesada.111
Duas iJAK são exploradas para o tratamento do vitiligo: ruxolitinibe 1,5% creme foi aprovado para uso nos EUA,112 e ritlecitinibe 10‐50mg/dia oral encontra‐se em estudo de fase IIb.113 Há relatos anedóticos do uso do tofacitinibe 5‐10mg/dia oral e tópico 2% creme ou pomada no vitiligo;114,115 entretanto, não foram publicados ensaios clínicos randomizados e controlados com essa substância.
O racional do uso de iJAK em vitiligo decorre de estudos farmacológicos pré‐clínicos que demonstraram a inibição da ação citolítica dos linfócitos T CD8+ e células NK quando inibidas enzimas da família das quinases (BTK, BMX, ITK, RLK e TEC) pelo ritlecitinibe, principalmente ITK, no caso dos linfócitos T citotóxicos, e RLK e TEC no caso das células NK. Ritlecitinibe é altamente seletivo para JAK3 e, potencialmente, inibe IL‐2 e IL‐15. Além disso, com base em estudos in vitro e ensaios celulares, ritlecitinibe pode diminuir a produção de IFN‐γ liberada por células T citotóxicas e NK, relacionadas a uma provável inibição de ITK.116,117
O ruxolitinibe é um inibidor de JAK1 e JAK2, cujo creme 1,5% diminuiu a concentração sérica de CXCL10, além de diminuir a ação pró‐inflamatória de quimiocinas e IL‐15 em cultivo celular de queratinócitos e melanócitos.118,119
O tofacitinibe é um inibidor de JAK1 e JAK3. Em um estudo com modelo murino de vitiligo‐induzido, o nível sérico de CXCL10 foi menor no grupo tratado com tofacitinibe oral comparado com o grupo placebo.120 Contudo, o ritlecitinibe tem maior efeito inibitório de JAK3, comparado ao tofacitinibe.117
Dois estudos randomizados, duplo‐cegos e controlados por placebo com ruxolitinibe creme a 1,5% envolveram pacientes maiores de 12 anos com vitiligo não segmentar e área afetada de no máximo 10% da superfície corporal. Os pacientes foram randomizados para usar o medicamento ou o veículo (2:1), 2×/dia na face ou no corpo por 24 semanas; depois desse período, todos utilizaram o ativo, totalizando 52 semanas. Os desfechos primários foram a melhora desde o início do tratamento de ao menos 75% do Vitiligo Area Score Index (VASI) facial, ou resposta F‐VASI75 (F‐VASI com escore de 0 a 3) na semana 24.112
No total, 674 pacientes foram incluídos, 330 em um estudo denominado TRue‐V1 e 344 no chamado TRue‐V2. No primeiro estudo, a porcentagem de pacientes que atingiy reposta F‐VASI75 na semana 24 foi de 29,8% no grupo ruxolitinibe e 7,4% no grupo placebo (risco relativo, 4,0; intervalo de confiança [IC] de 95%, 1,9‐8,4); no segundo estudo, 30,9% e 11,4%, respectivamente (risco relativo, 2,7; 95%IC 1,5‐4,9). Os EA mais comuns em cada estudo foram acne (6,3% e 6,6%), nasofaringite (5,4̈% e 6,1%) e prurido no local de aplicação (5,4% e 5,3%), respectivamente.
Um importante desfecho secundário foi a diminuição de 50% do T‐VASI (área total afetada), desde o início do tratamento (T‐VASI50). Aproximadamente 20% dos pacientes alcançaram uma resposta T‐VASI50 e 50% dos pacientes atingiram F‐VASI50. Essa desproporção é usual em ensaios de vitiligo em virtude da maior concentração de folículos na face, de onde migram melanoblastos para a repigmentação.121,122
Até o momento, não existem estudos randomizados e controlados com ruxolitinibe oral no tratamento do vitiligo, tampouco ruxolitinibe tópico comparado ou associado aos tratamentos ativos de escolha (fototerapia, inibidores de calcineurina ou corticosteroides tópicos).
Outro estudo randomizado controlado com placebo utilizou ritlecitinibe oral dose‐escalonado, por 24 semanas, com um período de extensão de 24 semanas e mais oito semanas de seguimento, em pacientes com vitiligo não segmentar entre 18 e 65 anos de idade, área corporal afetada entre 4% e 50%, e área facial afetada> 25%.113 Os pacientes foram randomizados em cinco grupos com ritlecitinibe e um com placebo. Dois grupos receberam dose de ataque de 100 ou 200mg/dia por quatro semanas seguidos de 50mg/dia de manutenção, por 20 semanas. Três grupos receberam 50, 30 ou 10mg/dia por 24 semanas, e um grupo recebeu placebo por 24 semanas. Os pacientes foram alocados no período de extensão com base na resposta na semana 16; não respondedores (< 50% T‐VASI) foram alocados para um grupo aberto com brepocitinibe, outro aberto com ritlecitinibe+fototerapia com UVB de banda estreita (NBUVB) ou ritlecitinibe 200mg de dose de ataque+50mg/dia. O desfecho primário foi a mudança desde o início no F‐VASI central na semana 24; o desfecho secundário foi a proporção de pacientes com F‐VASI75 central na semana 24.
Um total de 364 pacientes recebeu tratamento, e para fins de comparação com o estudo de ruxolitinibe tópico vamos usar os resultados do desfecho secundário. F‐VASI75 na semana 24 foram alcançados em 12,1%, 8,5%, 7,7%, 2,7%, 2,3% e 0% nos grupos ritlecitinibe 200/50mg, 100/50mg, 50mg, 30mg, 10mg e placebo, respectivamente. Quanto à segurança, 77% dos pacientes apresentaram algum EA, e os três mais comuns foram nasofaringite (15,9%), infecções de trato respiratório superior (11,5%) e cefaleia (8,8%).
Apesar dos resultados modestos na semana 24, percebeu‐se a aceleração da repigmentação após a semana 28. A morosidade no processo de repigmentação é bem fundamentado, o que justifica a fototerapia como tratamento adjuvante na aceleração da repigmentação, pelo menos com as medicações antitirosina quinase aqui apresentadas, como no caso do tofacitinibe, ruxolitinibe e mesmo pela alocação de alguns dos não respondedores com ritlecitinibe para um braço de ritlecitinibe associado com NBUVB.123,124
Se comparados os estudos com ruxolitinibe tópico e ritlecitinibe oral em relação ao F‐VASI75 na semana 24, a resposta do ruxolitinibe foi superior (30% vs. 12,1%).125 É relevante ainda que esse desfecho tenha sido atingido com a maior dose de ritlecitinibe, o F‐VASI tenha sido calculado apenas no centro da face e os pacientes do ruxolitinibe apresentavam fototipos mais baixos que os do estudo do ritlecitinibe, que são mais difíceis de repigmentar.113
Até o momento, não existem estudos randomizados e controlados com ruxolitinibe oral no tratamento do vitiligo, tampouco, ruxolitinibe tópico comparado ou associado aos tratamentos tópicos de escolha (inibidores de calcineurina, ou corticosteroides tópicos). Não obstante, em um estudo com camundongos como modelo, foi administrado oralmente tofacitinibe ou ruxolitinibe. Como resultado, foi observada superioridade do tofacitinibe em relação ao ruxolitinibe na diminuição nos escores de vitiligo dos pacientes afetados.116,126
Desse modo, os iJAK revelaram‐se potenciais terapêuticas para o vitiligo, com bom perfil de segurança, porém, que devem ser explorados em associação com outras medidas (p. ex., fototerapia) para maximizar sua eficácia.
Outras dermatosesA hidradenite supurativa (HS) decorre da desregulação do sistema imune inato e adaptativo, evolvendo diversas citocinas mediadas pela via JAK‐STAT (p. ex., IL‐17, IL‐23, IL‐10) e, em menor quantidade, TNF‐α. Tofacitinibe 5mg, 2×/dia, foi relatado em dois pacientes, com resultados favoráveis.127
Um estudo retrospectivo com 20 pacientes em uso de upadacitinibe demonstrou que 75% dos indivíduos atingiram HiSCR50 em quatro semanas, e 100% em 12 semanas. Todos receberam 15mg/dia, e os que não atingiram HiSCR50 em quatro semanas tiveram a dose aumentada para 30mg/dia.128 Estudos de fase II com INCB054707, 15mg, 30mg, 60mg e 90mg/dia, demonstraram que 43%, 56%, 56% e 88% dos pacientes,respectivamente, atingiram HiSCR na semana 8.129
Pioderma gangrenoso (PG) é dermatose inflamatória neutrofílica com incidência estimada de 3‐10 casos/milhão de pessoas‐ano. Sua patogênese é mal compreendida, mas parece envolver a desregulação tanto da imunidade inata quanto da adaptativa.130 A frequência de comorbidades imunomediadas associadas, superexpressão de citocinas inflamatórias, além da resposta a imunomoduladores (p. ex., corticoides, anti‐TNFα, inibidores de calcineurina) reforçam um mecanismo subjacente imunomediado.131
Não existe abordagem padronizada na estratégia terapêutica imunossupressora do PG, havendo casos resistentes.131,132 Já foram demonstradas desregulações da via JAK‐STAT no PG, com JAK1, JAK2, JAK3 e STAT1 a STAT6 superexpressas nas lesões. Mutações da via JAK‐STAT também foram implicadas na progressão do PG.130 Apesar de não haver estudos clínicos controlados, diversos relatos demonstraram benefícios do tofacitinibe, ruxolitinibe e baricitinibe em casos de PG.132–135Dermatomiosite (DM) é doença multissistêmica em que ocorre ativação do sistema imune inato com secreção de interferonas do tipo I. As atuais opções de tratamento incluem uma combinação de fotoproteção, glicocorticoides (GCs), imunossupressores e imunomoduladores.
Tofacitinibe foi relatado com sucesso em casos refratários de DM em doses de 5mg, 2×/dia, em 34 pacientes, 11mg/dia (liberação prolongada) em 10 pacientes e 10mg, 2×/dia, em dois pacientes. Ruxolitinibe foi usado em sete pacientes com dose máxima de até 30mg/dia. Após o início do tratamento, não ocorreu nenhuma recidiva em 3‐15 meses, havendo melhora dos sinais cutâneos, sintomas e testes de força muscular.136 No entanto, a maioria dos estudos incluídos nessa revisão foi de séries de casos, que são suscetíveis a viés de observação, e não houve comparação direta com outro método de tratamento.
A doença enxerto versus hospedeiro (DEVH) desenvolve‐se após transplante alogênico de células‐tronco hematopoéticas, quando as células T do doador não apenas reconhecem as células tumorais remanescentes como estranhas, mas também o tecido do receptor, levando a uma condição potencialmente grave. Órgãos‐alvo típicos incluem a pele, o fígado e os do trato digestório. Atualmente, todas as estratégias aprovadas para o tratamento da DEVH são terapias imunossupressoras, como a corticoterapia.
Como a DEVH é caracterizada principalmente pelo aumento das citocinas pró‐inflamatórias, esclerose sistêmica e dano de órgãos internos, a inibição da sinalização das tirosina quinases ativadas revelou‐se estratégia promissora para reduzir a gravidade e a progressão da doença. Várias pequenas moléculas estão sendo estudadas (fases I a III), entre elas: ruxolitinibe, itacitinibe, baricitinibe, ibrutinibe (inibidor da tirosina quinase de Bruton) e pacritinibe.
O tratamento com ruxolitinibe para DEVH aguda refratária à corticoterapia diminui os níveis de citocinas pró‐inflamatórias no soro dos pacientes e o número de células T ativadas, com taxa de resposta total de 82%. Efeitos similares foram obtidos em pacientes tratados com itacitinibe. Na DEVH crônica refratária, estudo de fase III com ruxolitinibe demonstrou quase o dobro de valores no alcance dos objetivos primários do tratamento em relação ao grupo controle.137
O reconhecimento dos iJAK como opção terapêutica para o tratamento de dermatoses pruriginosas crônicas começou a partir da DA. Porém, recentemente o gusacitinibe oral recebeu a designação de fast track pela FDA para o tratamento de eczema crônico das mãos. Além disso, iniciaram‐se ensaios clínicos do delgocitinibe tópico para tratamento de eczema crônico das mãos em pacientes pediátricos e adultos. Ainda, como a patogênese do prurigo nodularis (PN) envolve inflamação mediada por Th2 e Th17/Th22, que podem ser atenuadas por meio dos iJAK, estudos de fase II estão examinando o papel de abrocitinibe no tratamento do PN e prurido crônico de origem desconhecida.61
Conclusão e pesquisas futurasHá intensa atividade científica ligada ao desenvolvimento de medicamentos que interfiram em diferentes níveis da via JAK‐STAT, assim como a exploração de novas indicações e regimes posológicos.
O perfil de segurança da inibição das JAK, especialmente da JAK1 ou JAK3, associada à modulação da resposta imune, deve levar ao progressivo aumento do uso de iJAK em comparação aos imunossupressores (como os corticosteroides orais, azatioprina, micofenolato, metotrexato e ciclosporina) no tratamento de dermatoses inflamatórias e autoimunes. Todavia, apenas os ensaios clínicos com comparadores ativos podem posicionar os iJAK quanto às melhores indicações, regimes posológicos e seus aspectos farmacoeconômicos.
Por fim, a maior disponibilidade de moléculas deve levar à redução do custo ao sistema de saúde e aumentar o acesso aos pacientes. Em contrapartida, seu uso de longo prazo deve esclarecer aspectos ligados à manutenção da eficácia terapêutica, o impacto do bloqueio imunológico de vias seletivas de citocinas, além da segurança quanto à emergência de infecções e neoplasias malignas.
Suporte financeiroNenhum.
Contribuição dos autoresHélio Amante Miot: Idealização do estudo, escrita e aprovação do texto final.
Paulo Ricardo Criado: Idealização do estudo, escrita e aprovação do texto final.
Caio César Silva de Castro: Idealização do estudo, escrita e aprovação do texto final.
Mayra Ianhez: Idealização do estudo, escrita e aprovação do texto final.
Carolina Talhari: Idealização do estudo, escrita e aprovação do texto final.
Paulo Müller Ramos: Idealização do estudo, escrita e aprovação do texto final.
Conflito de interessesDr. Caio Castro: Advisory Board ‐ Sanofi, Aché, Sun‐Pharma, Galderma. Speaker ‐ Abbvie, Jansen, Novartis, Sanofi, Leo‐ Pharma. Pesquisa Clínica ‐ Abbvie, Pfizer, Jansen, Sanofi.
Dr. Paulo Criado pesquisa Clínica para Pfizer, Novartis, Sanofi e Lilly; Advisory board Pfizer, Galderma, Takeda, Hypera, Novartis, Sanofi. Palestrantre: Pfizer, Abbvie, Sanofi, Hypera, Takeda, Novartis.
Dr. Hélio Miot: Advisory Board – Johnson & Johnson, L’Oréal, Theraskin e Pfizer; pesquisa clínica Abbvie e Merz.
Dra. Mayra Ianhez: Speaker e Advisory Board ‐ Abbvie, Sanofi‐Genzyme, Pfizer, Janssen, Galderma, Theraskin.
Dr. Paulo Müller Ramos: Advisory Board – Pfizer;
Dra. Carolina Talhari: Nenhum conflito de interesses.
Como citar este artigo: Miot HA, Criado PR, Castro CC, Ianhez M, Talhari C, Ramos P. JAK‐STAT pathway inhibitors in dermatology. An Bras Dermatol. 2023;98:656–77.
Trabalho realizado no Departamento de Dermatologia da Faculdade de Medicina, Universidade Estadual Paulista, Botucatu, SP, Brasil.
