









As vasculites constituem um grupo de diversas condições clínicas nas quais o principal achado ao exame histopatológico é a necrose fibrinoide nas paredes dos vasos sanguíneos. Neste artigo são analisados os principais aspectos dermatológicos relevantes para o diagnóstico clínico e laboratorial de síndromes de vasculite cutânea e sistêmica de vasos de pequeno e médio porte. Também são discutidos os aspectos mais importantes do tratamento.
Na prática clínica diária, os dermatologistas são frequentemente solicitados a avaliar dois grupos de pacientes: o primeiro, que se apresenta com vasculites de pequenos vasos, e o segundo, que se apresenta com arterite cutânea (acometimento de vasos de médio calibre). Diante desse cenário, recomendamos a caracterização das manifestações dermatológicas no momento da apresentação inicial, bem como a avaliação dos achados de uma biópsia adequada, ou seja, que represente todas as camadas da pele afetadas, a fim de alcançar um diagnóstico adequado. O momento de sua feitura também é de valor substancial, uma vez que o exame pode ser não diagnóstico quando feito muito precoce ou tardiamente. Uma lesão recente, com até 24‐48 horas de aparecimento, é a mais adequada para o estudo histopatológico. Lesões com mais de 48 horas podem revelar, independentemente da vasculite subjacente, um infiltrado rico em linfócitos. A avaliação de dados adicionais, como aqueles fornecidos por exame de imunofluorescência direta (IFD), assim como pelo status do anticorpo anticitoplasma de neutrófilos (ANCA), estreitará ainda mais os possíveis diagnósticos diferenciais. A biópsia para IFD deve ser feita em uma lesão com até 8‐24 horas de surgimento. Caso contrário, os leucócitos degradam os complexos imunes e o resultado torna‐se falsamente negativo.1–4
Vasculite cutânea de pequenos vasos de órgão únicoA vasculite cutânea de pequenos vasos de órgão único é uma síndrome caracterizada por manifestações clínicas e histológicas de vasculite de pequenos vasos (VCPV) da pele sem envolvimento de vasos de qualquer outro órgão, o que pode ser confirmado pela anamnese, pelo exame físico e por exames laboratoriais selecionados.5 A maioria dos casos manifesta‐se como púrpura palpável ou máculas eritematosas. Contudo, urticária, vesículas hemorrágicas e úlceras rasas podem ocorrer (fig. 1).2,3 Metade dos pacientes apresenta lesões exclusivamente nos membros inferiores; membros superiores, tronco e região glútea também podem ser afetados.6 A doença costuma ser leve e autolimitada, com bom prognóstico. Muitos pacientes com vasculite cutânea de pequenos vasos de órgão único apresentam um único episódio, que geralmente resolve em poucas semanas. Recidivas ocorrem em cerca de 10%‐25% dos casos. De modo geral, sintomas sistêmicos como febre, mal‐estar, perda de peso e artralgia podem estar presentes em todas as síndromes de vasculite cutânea.7–9
 púrpura palpável e úlceras necróticas nos membros inferiores; (B) necrose de células endoteliais de vênulas da derme papilar superficial com deposição de fibrina, infiltrado de neutrófilos e leucocitoclasia.")
Os achados histológicos clássicos da vasculite cutânea de pequenos vasos de órgão único incluem um infiltrado predominantemente neutrofílico que acomete vênulas pós‐capilares, depósitos fibrinoides, edema endotelial e extravasamento de hemácias. Um infiltrado inflamatório misto pode ser observado, particularmente em lesões mais antigas.1,3 É importante destacar que esse padrão histológico não é exclusivo de alguma entidade em particular. Infecções, picadas de inseto, pioderma gangrenoso e mesmo biópsias obtidas do fundo de úlceras crônicas podem exibir os achados correspondentes a uma vasculite leucocitoclástica.2 Sempre que possível, uma segunda biópsia deve ser feita para IFD, cujos achados revelam predominantemente depósitos de complemento (C3), IgM e fibrina. Novamente, esses achados são típicos, mas não específicos de qualquer vasculite cutânea de pequenos vasos de órgão único. Desse modo, a correlação clínico‐patológica é obrigatória antes de se estabelecer um diagnóstico definitivo.4
Vasculites cutâneas localizadas: eritema elevatum diutinum e granuloma facialEritema elevatum diutinum (EED) e granuloma facial (GF) são vasculites cutâneas crônicas localizadas que afetam vasos dérmicos de pequeno calibre. No EED, as lesões apresentam‐se como pápulas, placas ou nódulos, bem delimitados, com coloração eritematosa a acastanhada, superfície lisa e distribuição relativamente simétrica, de curso crônico e duração em torno de 5 a 10 anos. Elas ocorrem preferencialmente na superfície extensora das extremidades, topografia altamente característica, sobretudo na pele sobre as articulações. Embora raramente, o EED pode se apresentar sob a forma de pápulas disseminadas nos membros superiores e inferiores, erupções verrucosas ou lesões extensas nodulares e fibróticas em palmas e plantas. As lesões podem desaparecer espontaneamente, sem deixar cicatrizes, porém áreas atróficas hiper ou hipopigmentadas podem ser vistas. Essa vasculite afeta mais comumente pacientes do sexo feminino em qualquer faixa etária, embora seja mais frequente entre a quarta e a sexta décadas de vida.10–12
Embora a fisiopatologia do EED não seja totalmente compreendida, acredita‐se que as lesões cutâneas sejam causadas pelo depósito de complexos imunes nos pequenos vasos dérmicos, o que resulta em fixação do complemento e subsequente inflamação. Diversas publicações relacionam o EED a anormalidades hematológicas, particularmente gamopatias monoclonais por IgA; infecções pelo vírus da imunodeficiência humana (HIV), hepatite C (HCV), hepatite B (HBV) ou tuberculose; doenças autoimunes, como artrite reumatoide, policondrite recidivante e doença inflamatória intestinal; e tumores sólidos de pulmão ou mama.13 ANCA de classe IgA pode ser um relevante marcador da doença. A imunoeletroforese deve ser usada para investigar possíveis gamopatias associadas.11,12 O EED pode cursar com alterações oculares, inclusive esclerite nodular, panuveíte e queratite periférica.14
O GF apresenta predileção por indivíduos de meia‐idade, sobretudo do sexo masculino, e é caracterizado pelo aparecimento de pápulas, placas ou nódulos purpúricos a acastanhados, assintomáticos ou levemente pruriginosos, normalmente localizados na face. Sua superfície é lisa e pode levar à acentuação dos óstios foliculares. Existe uma variante que afeta a mucosa nasal, denominada fibrose angiocêntrica eosinofílica, a qual raramente coexiste com lesões cutâneas faciais. O envolvimento extrafacial é excepcional, mas pode ocorrer em áreas fotoexpostas ou na genitália. Embora usualmente refratário ao tratamento e de rara resolução espontânea, o GF não tem sido associado a doenças sistêmicas. Seu diagnóstico diferencial: sarcoidose, linfomas e pseudolinfomas cutâneos, lúpus discoide, hanseníase, rosácea granulomatosa e infiltrado linfocítico de Jessner.15,16
EED e GF são vasculites fibrosantes sem sinais de reação granulomatosa. Lesões antigas demonstram um aspecto de fibrose concêntrica “em casca de cebola” ao redor dos vasos.11 Histologicamente, no GF existe um infiltrado inflamatório polimórfico dérmico denso, superficial e profundo, em sua maioria perivascular. Caracteristicamente, um infiltrado composto por todos os tipos de células inflamatórias, inclusive neutrófilos, linfócitos, plasmócitos e eosinófilos, encontra‐se separado da epiderme sobrejacente por uma estreita zona de Grenz. Enquanto isso, no EED não se observa achado histológico específico que o diferencie de outras vasculites leucocitoclásticas. Nem sempre a epiderme é poupada e não se observa zona de Grenz.11,14 As formas vesicobolhosa, hemorrágica, ulcerativa e anular de EED são pouco usuais. Depósitos de IgA no nível das vesículas são detectados no EED vesicobolhoso, uma variante cujo diagnóstico diferencial mais relevante é a dermatite herpetiforme.17
Urticária vasculiteA urticária vasculite (UV) é caracterizada por episódios recorrentes de urticária com duração maior do que 24‐36 horas, com características histopatológicas de vasculite leucocitoclástica que envolve principalmente vênulas pós‐capilares.18 Essa vasculite tem uma prevalência aumentada em pacientes do sexo feminino, com pico na quarta década de vida, e é extremamente rara em crianças.19 A UV pode ser idiopática (mais frequentemente) ou estar associada a reações medicamentosas e doenças crônicas, como infecções, malignidades, doenças hematológicas e patologias do tecido conectivo, particularmente lúpus eritematoso sistêmico (LES) e síndrome de Sjögren. Diferentemente da urticária crônica idiopática, as lesões de UV cursam com dor, sensibilidade e calor local, apresentam uma região central de coloração rubra a acastanhada à diascopia e podem evoluir com hiperpigmentação residual. Outras lesões cutâneas, como púrpura ou necrose, são eventualmente observadas.18,19 Não apenas os membros inferiores, mas qualquer segmento corporal pode ser acometido.20 Angioedema é observado em menos da metade dos pacientes. A maioria dos casos segue um curso crônico benigno, com duração de 3‐4 anos, e responde mal aos anti‐histamínicos.19
A UV divide‐se fundamentalmente em: (i) forma normocomplementêmica, responsável por 80% dos casos, um distúrbio leve, autolimitado e com envolvimento cutâneo predominante; (ii) síndrome vasculite urticariforme hipocomplementêmica, uma condição rara e potencialmente fatal com hipocomplementenemia persistente por pelo menos seis meses. Essa forma está geralmente associada a manifestações sistêmicas, como artrite e artralgia/artropatia de Jaccoud, bem como à proteinúria e hematúria devido à glomerulonefrite em até 50% dos pacientes; doença pulmonar obstrutiva crônica, a principal causa de óbito entre os pacientes com UV; conjuntivite, uveíte ou episclerite; sintomas gastrintestinais; e doença cardiovascular.21,22
A suspeita clínica de UV ocorre diante de quadros de lesões urticariformes com apresentação atípica e exames laboratoriais sugestivos, tais como velocidade de hemossedimentação (VHS) elevada, em 42,6% dos pacientes; positividade para anticorpos antinucleares (ANA), em 33,4% dos pacientes; baixos níveis de C3, C4 e CH50, em cerca de um terço dos pacientes, sobretudo nas formas mais graves da doença; e presença de anticorpos anti‐C1q e/ou C1q supresso, em 55% dos pacientes com síndrome vasculite urticariforme hipocomplementêmica. Porém, o diagnóstico sempre precisa ser confirmado por meio de uma biópsia cutânea. Baixos níveis de C1q também podem ser encontrados no LES (61%), artrite reumatoide (20%), esclerodermia (15%), síndrome de Sjögren (15%), doença mista do tecido conjuntivo (15%) e infeção crônica pelo HCV (38%).15,19,23 A síndrome vasculite urticariforme hipocomplementêmica assemelha‐se ao LES clínica e imunologicamente e é considerada por alguns autores uma síndrome associada ao LES.24
Vasculite por IgAA vasculite por IgA (IgAV) é uma vasculite leucocitoclástica sistêmica de pequenos vasos que acomete arteríolas, capilares e, sobretudo, vênulas e está associada à deposição de IgA (isolada ou predominante). As vasculites são incomuns na faixa etária pediátrica, com exceção da doença de Kawasaki e da IgAV, essa responsável por mais da metade de todos os casos de vasculites em crianças e notavelmente mais comum entre os 3 e 15 anos. A IgAV frequentemente ocorre cerca de 10 dias após uma condição inflamatória aguda. Por muito tempo, as infecções por estreptococos do grupo B foram consideradas o único fator causal associado à IgAV. Contudo, atualmente, sabe‐se que elas correspondem a menos de um terço de todos os casos. Não apenas uma ampla variedade de agentes infecciosos, como vírus, bactérias e talvez protozoários, mas também picadas de himenópteros, medicamentos e imunização podem desencadear IgAV.7 Aproximadamente 25% dos casos ocorrem em adultos, os quais apresentam achados semelhantes àqueles vistos em crianças. No entanto, embora os adultos raramente tendam à intussuscepção intestinal, apresentam risco mais elevado de envolvimento renal importante. Particularmente nessa faixa etária, o IgAV pode ocorrer durante a evolução ou antes do diagnóstico de malignidade.6,7
Uma erupção cutânea que afeta simetricamente extremidades inferiores e região glútea é um sinal inequívoco de IgAV em crianças. Lesões menos intensas podem acometer a face, o tronco, os antebraços e os punhos. Habitualmente, apresentam margens bem definidas, variam em tamanho de alguns milímetros a poucos centímetros, não desaparecem à diascopia e tipicamente ocorrem em surtos. Elas também podem coalescer, formar grandes áreas de envolvimento cutâneo. Lesões purpúricas geralmente desaparecem dentro de um mês, evoluem com hiperpigmentação acastanhada por mais duas semanas. Edema localizado e não depressível é observado em cerca de 50% dos casos de IgAV. Ele é geralmente indolor e envolve mais de uma área, normalmente a região dorsal das mãos, tornozelos e pés. Com menor frequência, os tecidos periorbitais, lábios, fronte e couro cabeludo são afetados.7
Várias outras manifestações e achados atípicos podem ser identificados na IgAV. Eles incluem Köbnerization nos membros inferiores, região inguinal, cintura abdominal, punhos ou antebraços; fenômeno de fragilidade capilar de Rumpel‐Leede, que consiste no aparecimento de púrpuras palpáveis após a aplicação de um manguito de pressão arterial no braço, em um ponto intermediário entre a pressão sistólica e a pressão diastólica, por 5 a 10 minutos; e envolvimento uni ou bilateral da bolsa escrotal, caracterizado por coloração eritematosa, edema e moderada sensibilidade local. Vesículas e bolhas podem ocorrer em até duas semanas após o surgimento das primeiras lesões cutâneas. Em 5%‐10% dos casos, os achados cutâneos persistem por mais de dois meses. Do mesmo modo, as recorrências, definidas pelo reaparecimento de manifestações dermatológicas após um período de quatro ou mais semanas de recuperação completa, ocorrem em 5%‐10% dos pacientes.7 O acompanhamento rigoroso, com análise de sedimento urinário e monitoramento da pressão arterial, deve ser mantido durante pelo menos seis meses, tendo em vista que o envolvimento renal na IgAV é tipicamente leve e assintomático.4
Além da análise de sedimento urinário, a avaliação laboratorial inicial deve incluir a dosagem de ureia, creatinina e eletrólitos no sangue, hemograma completo, coagulograma e VHS. Como na doença de Kawasaki e em muitas outras condições inflamatórias, trombocitose reativa pode ser observada. Os níveis de complemento geralmente são normais, assim como ANA e ANCA são usualmente negativos. O achado de depósitos de IgA em pequenos vasos à IFD é sensível, mas não específico da IgAV, uma vez que pode ser detectado nas vasculites associadas a doenças sistêmicas, como LES, crioglobulinemia, paraproteinemia/gamopatia, eritema nodoso, estase venosa, vasculopatias secundárias a coagulopatias e vasculopatia livedoide.4,25 Até o momento, nenhum tratamento mostrou‐se capaz de reduzir a duração da doença.4 Alguns autores classificam a síndrome de Finkelstein‐Seidlmayer como uma entidade distinta da IgAV, dado que a primeira apresenta envolvimento cutâneo isolado e que depósitos de IgA são encontrados em não mais do que um terço dos casos.7
Várias classificações para o diagnóstico de IgAV foram propostas ao longo dos anos. Em 2010, os critérios propostos pela Liga Europeia contra o Reumatismo (EULAR) /PRINTO/PRES foram formalmente publicados. Púrpuras palpáveis não trombocitopênicas, frequentemente agrupadas, normalmente em membros inferiores, são um achado obrigatório. Além disso, os pacientes afetados precisam apresentar ao menos um dos seguintes: dor abdominal difusa; artrite ou artralgia; comprometimento renal (proteinúria>0,3 g/24h ou relação de albumina/creatinina>30 mmoL/mg em amostra de urina pela manhã; e/ou hematúria>cinco hemácias por campo de grande aumento ou duas [+] ou mais por tira reativa ou cilindros hemáticos no sedimento urinário) e biópsia com vasculite leucocitoclástica típica ou glomerulonefrite proliferativa, com depósito predominante de IgA.25 A escala CAAR varia de 0 a 3, em que 0 indica ausência, 1 envolvimento leve, 2 moderado e 3 grave, no tocante ao envolvimento cutâneo, abdominal, articular e renal. O índice de atividade da doença pode ser calculado somando a pontuação correspondente a cada um dos quatro itens (tabela 1).7
Avaliação do acometimento na vasculite por IgA, com base na classificação CAAR para sinais e sintomas cutâneos, abdominais, articulares e renais
| Envolvimentocutâneo | Ausente: sem lesões cutâneas; |
| Moderado: lesões na pele localizadas (a) nas nádegas e extremidades inferiores e (b) no tronco ou nas extremidades superiores; | |
| Grave: lesões cutâneas localizadas em (a) glúteos e extremidades inferiores, (b) tronco e (c) extremidades superiores. | |
| Envolvimento abdominal | Ausente: sem sintomas, sem achados; |
| Leve: dor abdominal leve (induzida pelo exame físico médico); | |
| Moderado: dor abdominal moderada (queixas transitórias trazidas à consulta médica); | |
| Grave: dor abdominal intensa e/ou melena e/ou hematêmese e/ou intussuscepção. | |
| Envolvimento articular | Ausente: sem sintomas, sem achados; |
| Leve: sintomas ou achados do envolvimento articular, mas sem anormalidades funcionais; | |
| Moderado: sintomas e achados do envolvimento articular que causam redução funcional moderada (por exemplo, claudicação); | |
| Grave: sintomas e achados que causam perda funcional moderada (por exemplo, incapacidade de andar). | |
| Envolvimento renal | Ausente: exame do sedimento urinário normal. |
| Leve: hematúria patológica, proteinúria normal (bastões negativos ou [+]). | |
| Moderado: hematúria patológica, proteinúria leve a moderada (bastão+a [++]). | |
| Grave: hematúria patológica, proteinúria grave (bastão ≥ [+++]). |
A classificação CAAR para envolvimento cutâneo, abdominal, articular e renal na vasculite por IgA varia de 0 a 3, em que 0 indica ausência, 1 doença leve, 2 moderada e 3 grave. A atividade da doença pode ser calculada pela soma dos quatro itens.
A vasculite eosinofílica cutânea recorrente é uma vasculite necrosante cutânea de pequenos vasos dérmicos raros. Ao exame histopatológico, apresenta infiltrado inflamatório quase exclusivamente eosinofílico, com poucos neutrófilos perivasculares. Pode manifestar‐se por meio de placas urticariformes anulares, pápulas purpúricas pruriginosas, angioedema ou lesões necróticas. Não se observam manifestações sugestivas de doença sistêmica. A doença segue um curso recorrente e responde prontamente aos corticosteroides sistêmicos.26
As proteínas citotóxicas dos grânulos de eosinófilos depositam‐se próximas aos vasos sanguíneos, sugerem que essas células medeiam o dano vascular. Os eosinófilos liberam interleucina (IL)‐5, C4 e fator ativador de plaquetas, o que pode levar ao aumento da permeabilidade vascular. Não se observa deposição de Ig ao longo das paredes dos vasos à IFD. A vasculite eosinofílica cutânea recorrente geralmente está associada à eosinofilia no sangue periférico, que é uma característica comum a muitas doenças, como síndrome hipereosinofílica, celulite eosinofílica, granulomatose eosinofílica com poliangeíte (GEPA) e fasciíte eosinofílica, embora ela nem sempre se correlacione com a gravidade da doença. Particularmente na síndrome hipereosinofílica e na celulite eosinofílica observa‐se infiltração eosinofílica em torno dos vasos dérmicos, porém não há vasculite necrotizante verdadeira.26
Edema hemorrágico agudo da infânciaO edema hemorrágico agudo da infância (EHAI) tem sido descrito na literatura sob uma variedade de sinônimos, inclusive “púrpura de Henoch‐Schönlein da primeira infância”, “eritema multiforme infantil” e “urticária vasculite da infância”. Classicamente, caracteriza‐se pela tríade de lesões purpúricas palpáveis, edema e febre. Envolvimento de membranas mucosas é raro. Essa vasculite é mais frequente no sexo masculino, apresenta distribuição etária que varia entre 2 e 60 meses, embora 80% dos casos ocorram em crianças de 6 a 24 meses. Em quase 75% dos casos, está temporariamente associada, em até duas semanas, a uma variedade de infecções virais ou bacterianas e imunização. Embora sua etiologia permaneça desconhecida, acredita‐se que o EHAI seja mediado por complexos imunes.3,27
Até o momento, nenhum critério de classificação foi proposto. O diagnóstico geralmente é feito clinicamente, por meio de cinco sintomas e sinais: (i) idade de até 2 anos; (ii) lesões cutâneas típicas: em alvo, acometem predominantemente a face sobre regiões malares, orelhas e as extremidades, poupam o tronco; edema da face, orelhas e extremidades, sem prurido ou arranhões; (iii) estado geral preservado, sem cianose ou palidez, sem diminuição de temperatura nas extremidades e com enchimento capilar inferior a dois segundos, ausência de hipo ou hiperventilação; (iv) envolvimento articular ou abdominal ausente; e (v) recuperação espontânea dentro de 2‐3 semanas.3
O envolvimento visceral é raro, embora artrite, sangramento gastrintestinal e edema escrotal sejam descritos. Em casos duvidosos, a biópsia de pele pode ser de grande valor para o diagnóstico. À histologia, observam‐se os achados típicos de uma vasculite leucocitoclástica, com ou sem necrose fibrinoide. A IFD pode revelar depósitos de C3, fibrinogênio e IgG e, menos comumente, IgM ou IgE. Deposição de IgA tem sido vista em um terço dos casos. Diante do seu curso benigno e autolimitado, com remissão espontânea completa em uma a três semanas, e dado que o EHAI usualmente não apresenta boa resposta aos corticosteroides, a maioria dos autores defende uma abordagem conservadora, exceto para os casos que evoluem com inflamação severa, como aqueles com inflamação escrotal.27
Vasculites mistas, predominantemente de pequenos e médios vasosVasculites associadas ao ANCAAs vasculites associadas ao ANCA (VAA) podem estar limitadas a um órgão, sobretudo pulmões e rins, ou afetar vários sistemas desde a sua apresentação inicial. Qualquer tipo de vaso pode ser acometido, inclusive capilares, vênulas e arteríolas. Dentre as VAA estão incluídas a GEPA, a poliangeíte microscópica (PAM), a granulomatose com poliangeíte (GPA) e as VAA induzidas por drogas, todas exibem uma ampla variedade de manifestações cutâneas que podem coexistir em um mesmo paciente (fig. 2). Portanto, o diagnóstico deve basear‐se em uma história médica detalhada, exame físico preciso, análise histopatológica da biópsia da pele e status do ANCA.28–30
 granulomatose eosinofílica com poliangeíte manifesta‐se como púrpuras palpáveis e petéquias, ambas manifestações comuns e inespecíficas desse grupo de vasculites; (B) um paciente portador de poliangeíte microscópica com úlceras necróticas extensas bilateralmente nos membros inferiores.")
Vasculites associadas ao ANCA: (A) granulomatose eosinofílica com poliangeíte manifesta‐se como púrpuras palpáveis e petéquias, ambas manifestações comuns e inespecíficas desse grupo de vasculites; (B) um paciente portador de poliangeíte microscópica com úlceras necróticas extensas bilateralmente nos membros inferiores.
Com a técnica de imunofluorescência indireta (IFI), três padrões principais de ANCA foram descritos: citoplasmático (c)‐ANCA, presente na maioria dos pacientes com GPA ativa; perinuclear (p)‐ANCA, observado em pacientes com PAM e GEPA; e atípico (a)‐ANCA, mais raro. Os ANCA podem estar relacionados à exposição a medicamentos, particularmente propiltiouracil, hidralazina e cocaína. A presença e especificidade antigênica dos ANCA fornecem informações clínicas potencialmente relevantes: pacientes com c‐ANCA ou p‐ANCA apresentam comprometimento de sistemas orgânicos distintos, assim como diferentes padrões de resposta à terapia medicamentosa de indução (prescrita para pacientes com a forma ativa da doença) e risco de recidiva. Entretanto, sua simples presença não é confirmatória para a existência de uma vasculite sistêmica, tendo em vista que alguns casos de vasculite cutânea de órgão único apresentam ANCA positivo e que níveis baixos desses autoanticorpos podem ser encontrados em diversos estados inflamatórios sistêmicos e doenças pulmonares.28–30
A GEPA acomete pacientes de ambos os sexos, com uma média de idade ao diagnóstico de 50,3±15,7 anos. Asma pode preceder a vasculite sistêmica em até 20 anos. O aparecimento de sinusite/polipose nasal/rinite (58,8%); mononeurite multiplex (46%); lesões cutâneas (39,7%), sobretudo púrpuras palpáveis, rash urticariforme e nódulos subcutâneos tipicamente localizados nos membros e couro cabeludo, mas também de livedo e lesões necróticas, além de infiltrados pulmonares (38,6%), reforça a suspeita de GEPA. Envolvimento cardíaco, gastrintestinal e renal são encontrados em 27,4%; 23,2%; e 21,7% dos pacientes, respectivamente.31,32 Ocasionalmente, os achados iniciais podem ser bastante distintos dos padrões usualmente reconhecidos e essa complexidade pode contribuir para retardar seu diagnóstico e tratamento. As possíveis manifestações dermatológicas da GEPA variam, na literatura, de erupção eritema multiforme‐símile a lesões crônicas liquenificadas, previamente diagnosticadas como prurigo nodular.28 Ademais, embora a GEPA seja uma vasculite associada à presença do ANCA, em torno de 50% dos pacientes testam negativos para tais autoanticorpos.32
Na PAM, as lesões cutâneas são encontradas em 30%‐60% dos pacientes e ocorrem como manifestação inicial em 15%‐30% dos casos. Púrpuras palpáveis são comuns (≥ 75%); todavia, a doença também pode se apresentar com livedo, nódulos, isquemia digital e urticária.33 Nódulos e placas EED‐símile já foram descritos. Assim como na poliarterite nodosa (PAN), as manifestações dermatológicas estão frequentemente associadas a artralgia e comprometimento ocular.34 O envolvimento renal está presente em quase 80% dos pacientes e é caracterizado por glomerulonefrite rapidamente progressiva. Em relação ao envolvimento pulmonar, a apresentação clássica é de uma hemorragia alveolar difusa devido à capilarite.33 De modo geral, a PAM evolui de maneira rápida, mas há uma variante lentamente progressiva. A ausência de achados tais como microaneurismas e/ou estenose à angiografia e p‐ANCA positivo (50%‐65% no momento do diagnóstico) deve ser usada como critério para o diagnóstico de PAM. No entanto, alguns pacientes apresentam positividade para c‐ANCA.33
De acordo com o Consenso da Conferência de Chapel Hill de 2012 (CHCC2012), a GPA é definida como uma vasculite necrotizante da pele, rins, trato respiratório superior e inferior.35 Todavia, nos estágios iniciais os pacientes podem apresentar apenas uma ou duas das manifestações da tríade e aproximadamente 20% dos casos podem ser c‐ANCA negativo.36,37 O envolvimento do seio nasal é o achado mais comum e pode ser o único observado na forma localizada da doença.37 Acomete os pulmões em cerca de 50%‐90% dos pacientes, causa hemorragia alveolar e/ou nódulos parenquimatosos. Lesões cutâneas (10%‐50% dos casos) são mais frequentes naqueles com doença multiorgânica ativa e grave.36,37 Elas incluem púrpuras palpáveis; lesões polimórficas, entre elas pápulas e nódulos necróticos localizados sobre superfícies extensoras, comumente cotovelos; úlceras pioderma gangrenoso‐símile, também chamadas de pioderma maligno; úlceras em mucosa oral; hiperplasia gengival/gengivite em morango; e lesões necróticas do pênis.36,37 A atividade da doença apresenta correlação com os níveis de ANCA. A taxa de mortalidade, assim como a frequência de recidivas, é elevada em todas as VAA.33
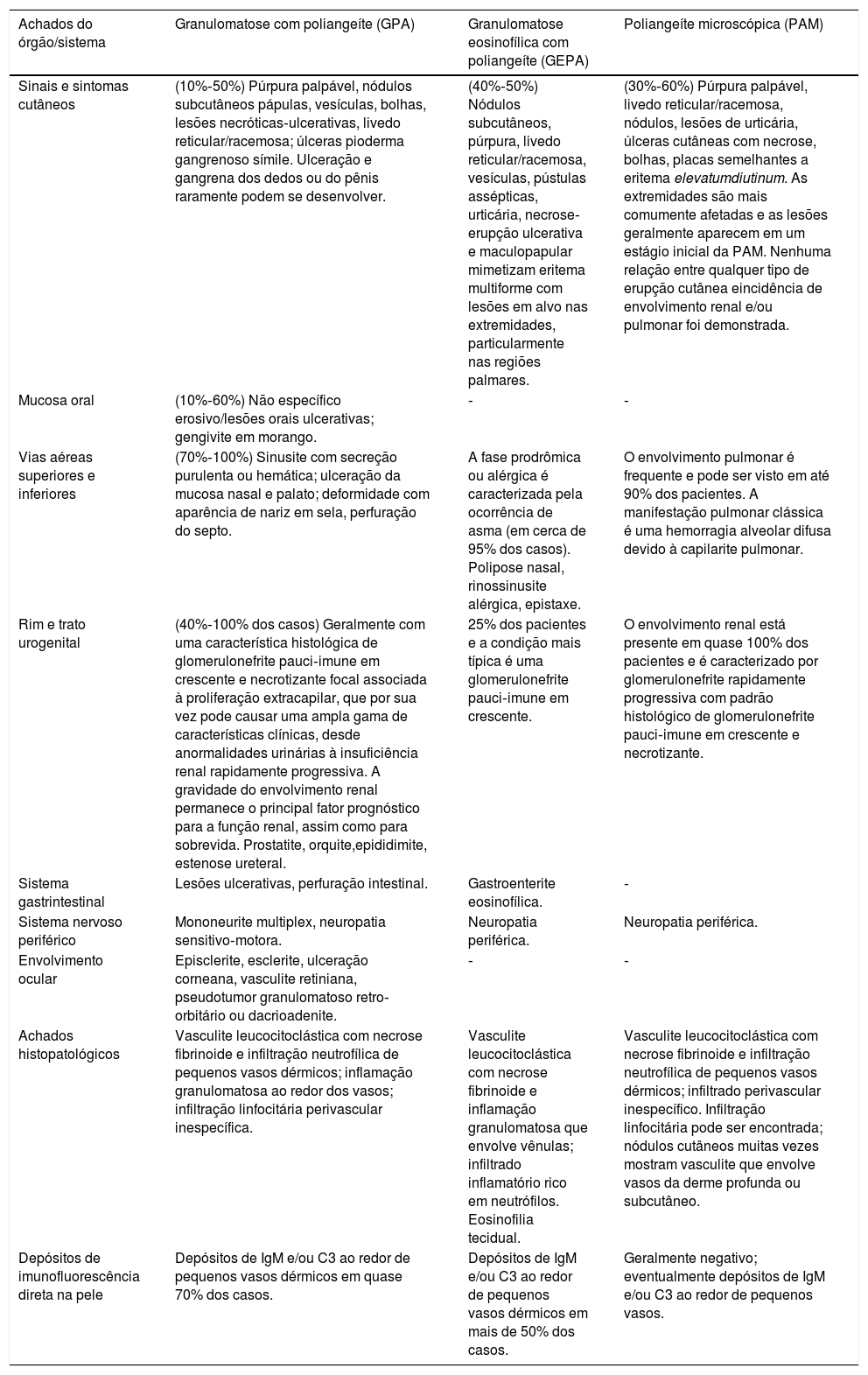
Histologicamente, as VAA manifestam‐se como vasculite neutrofílica de pequenos vasos. O status do c‐ANCA/p‐ANCA auxilia na interpretação dos achados da biópsia cutânea pelo dermatopatologista. Frequentemente, as VAA são chamadas de “pauci‐imunes”, termo que se refere à ausência de deposição de imunocomplexos nos vasos lesados. Na literatura, GEPA e GPA, mas não a PAM, são classificadas como vasculites granulomatosas, grupo que compreende ainda duas vasculites de grandes vasos, a arterite de células gigantes (temporal) e a arterite de Takayasu.28–30,34,38 Na tabela 2, adaptada de Marzano et al.,28 resumimos os achados cutâneos, sistêmicos e laboratoriais observados nas VAA.
Achados das vasculites associadas ao ANCA (anticorpos contra o citoplasma de neutrófilos)
| Achados do órgão/sistema | Granulomatose com poliangeíte (GPA) | Granulomatose eosinofílica com poliangeíte (GEPA) | Poliangeíte microscópica (PAM) |
|---|---|---|---|
| Sinais e sintomas cutâneos | (10%‐50%) Púrpura palpável, nódulos subcutâneos pápulas, vesículas, bolhas, lesões necróticas‐ulcerativas, livedo reticular/racemosa; úlceras pioderma gangrenoso símile. Ulceração e gangrena dos dedos ou do pênis raramente podem se desenvolver. | (40%‐50%) Nódulos subcutâneos, púrpura, livedo reticular/racemosa, vesículas, pústulas assépticas, urticária, necrose‐erupção ulcerativa e maculopapular mimetizam eritema multiforme com lesões em alvo nas extremidades, particularmente nas regiões palmares. | (30%‐60%) Púrpura palpável, livedo reticular/racemosa, nódulos, lesões de urticária, úlceras cutâneas com necrose, bolhas, placas semelhantes a eritema elevatumdiutinum. As extremidades são mais comumente afetadas e as lesões geralmente aparecem em um estágio inicial da PAM. Nenhuma relação entre qualquer tipo de erupção cutânea eincidência de envolvimento renal e/ou pulmonar foi demonstrada. |
| Mucosa oral | (10%‐60%) Não específico erosivo/lesões orais ulcerativas; gengivite em morango. | ‐ | ‐ |
| Vias aéreas superiores e inferiores | (70%‐100%) Sinusite com secreção purulenta ou hemática; ulceração da mucosa nasal e palato; deformidade com aparência de nariz em sela, perfuração do septo. | A fase prodrômica ou alérgica é caracterizada pela ocorrência de asma (em cerca de 95% dos casos). Polipose nasal, rinossinusite alérgica, epistaxe. | O envolvimento pulmonar é frequente e pode ser visto em até 90% dos pacientes. A manifestação pulmonar clássica é uma hemorragia alveolar difusa devido à capilarite pulmonar. |
| Rim e trato urogenital | (40%‐100% dos casos) Geralmente com uma característica histológica de glomerulonefrite pauci‐imune em crescente e necrotizante focal associada à proliferação extracapilar, que por sua vez pode causar uma ampla gama de características clínicas, desde anormalidades urinárias à insuficiência renal rapidamente progressiva. A gravidade do envolvimento renal permanece o principal fator prognóstico para a função renal, assim como para sobrevida. Prostatite, orquite,epididimite, estenose ureteral. | 25% dos pacientes e a condição mais típica é uma glomerulonefrite pauci‐imune em crescente. | O envolvimento renal está presente em quase 100% dos pacientes e é caracterizado por glomerulonefrite rapidamente progressiva com padrão histológico de glomerulonefrite pauci‐imune em crescente e necrotizante. |
| Sistema gastrintestinal | Lesões ulcerativas, perfuração intestinal. | Gastroenterite eosinofílica. | ‐ |
| Sistema nervoso periférico | Mononeurite multiplex, neuropatia sensitivo‐motora. | Neuropatia periférica. | Neuropatia periférica. |
| Envolvimento ocular | Episclerite, esclerite, ulceração corneana, vasculite retiniana, pseudotumor granulomatoso retro‐orbitário ou dacrioadenite. | ‐ | ‐ |
| Achados histopatológicos | Vasculite leucocitoclástica com necrose fibrinoide e infiltração neutrofílica de pequenos vasos dérmicos; inflamação granulomatosa ao redor dos vasos; infiltração linfocitária perivascular inespecífica. | Vasculite leucocitoclástica com necrose fibrinoide e inflamação granulomatosa que envolve vênulas; infiltrado inflamatório rico em neutrófilos. Eosinofilia tecidual. | Vasculite leucocitoclástica com necrose fibrinoide e infiltração neutrofílica de pequenos vasos dérmicos; infiltrado perivascular inespecífico. Infiltração linfocitária pode ser encontrada; nódulos cutâneos muitas vezes mostram vasculite que envolve vasos da derme profunda ou subcutâneo. |
| Depósitos de imunofluorescência direta na pele | Depósitos de IgM e/ou C3 ao redor de pequenos vasos dérmicos em quase 70% dos casos. | Depósitos de IgM e/ou C3 ao redor de pequenos vasos dérmicos em mais de 50% dos casos. | Geralmente negativo; eventualmente depósitos de IgM e/ou C3 ao redor de pequenos vasos. |
C, complemento; EED, eritema elevatum diutinum; GEPA, granulomatose eosinofílica com poliangeíte; GPA, granulomatose com poliangeíte; Ig, imunoglobulina; MPA, poliangeíte microscópica.
Adaptada de Marzano et al.28
A poliarterite nodosa sistêmica (PAN) é uma vasculite necrotizante que tipicamente afeta artérias musculares de médio calibre.39 De acordo com a definição proposta pelo CHCC2012, na PAN não há acometimento de vênulas pós‐capilares.35 Essa entidade tem sido historicamente dividida em dois subtipos principais: PAN sistêmica e PAN cutânea, atualmente denominada arterite cutânea. Os critérios de classificação do American College of Rheumatology (ACR) de 1990 para PAN do adulto e os critérios de classificação da Liga Europeia contra o Reumatismo (EULAR) para PAN infantil estão resumidos na tabela 3. Em relação às manifestações cutâneas, apenas livedo reticular é considerado um critério diagnóstico pelo ACR, ao passo que todos os tipos de lesões foram incluídos pelo EULAR. A PAN sistêmica é uma doença potencialmente fatal. Já a arterite cutânea é uma condição benigna crônica, com curso recidivante. A possibilidade de progressão da variante exclusivamente cutânea para a forma sistêmica é controversa; se ocorrer, provavelmente é um evento incomum.39–42
Critérios de classificação para poliarterite nodosa sistêmica em adultos (1990 American College of Rheumatology) e para poliarterite nodosa infantil (European League Against Rheumatism – EULAR)
| 1990 American College of Rheumatology (ACR)Critérios de classificação para PAN do adulto | A poliarterite nodosa é diagnosticada se pelo menos três desses 10 critérios estiverem presentes. A presença de três ou mais critérios produz uma sensibilidade de 82,2% e uma especificidade de 86,6%. | 1. Perda de peso> 4 kg; |
| 2. Livedo reticular; | ||
| 3. Dor ou sensibilidade testicular (geralmente unilateral, secundária à vasculite da artéria testicular, exige tratamento de emergência devido ao risco de isquemia irreversível); | ||
| 4. Mialgias, fraqueza ou sensibilidade nas pernas; | ||
| 5. Mononeuropatia ou polineuropatia; | ||
| 6. PA diastólica> 90 mmHg; | ||
| 7. Ureia elevada> 40mg/dL ou creatinina> 1,5 mg/dL, exceto por desidratação ou obstrução; | ||
| 8. Presença de antígeno ou anticorpo de superfície da hepatite B no soro; | ||
| 9. Arteriograma que mostre aneurismas ou oclusões das artérias viscerais, exceto por arteriosclerose, displasia fibromuscular ou outras causas não inflamatórias; | ||
| 10. Biópsia de pequena ou média artéria contendo PMN. | ||
| European League Against Rheumatism (EULAR)Critérios de classificação para PAN infantil | Doença sistêmica caracterizada por presença de uma biópsia que mostra vasculite necrotizante de pequenos e médios vasos ou anormalidades angiográficas (aneurismas ou oclusões) (critérios obrigatórios) (deve incluir angiografia convencional se a angiografia por ressonância magnética for negativa), mais pelo menos dois dos seguintes sete critérios: | 1. Envolvimento cutâneo (livedo reticular, nódulos subcutâneos sensíveis, outras lesões vasculíticas); |
| 2. Mialgia ou sensibilidade muscular; | ||
| 3. Hipertensão sistêmica, relativa aos dados normativos da infância; | ||
| 4. Mononeuropatia ou polineuropatia; | ||
| 5. Análise anormal da urina e/ou comprometimento da função renal (taxa de filtração glomerular<50% do normal para a idade); | ||
| 6. Dor ou sensibilidade testicular; | ||
| 7. Sinais ou sintomas sugestivos de vasculite de qualquer outro órgão importante (sistema nervoso central, gastrintestinal, cardíaco ou pulmonar). |
PA, pressão arterial; PAN, poliarterite nodosa; PMN, neutrófilos polimorfonucleares.
A PAN afeta mais frequentemente o sexo masculino e ocorre em todos os grupos étnicos. A idade média ao início da doença é de aproximadamente 50 anos e o pico de incidência ocorre nas quinta a sexta décadas de vida.34,43 A patogênese da PAN ainda não é completamente compreendida, embora a resposta clínica à terapia imunossupressora corrobore o conceito de que os mecanismos que envolvem os sistemas imunológicos inato e adaptativo desempenhem um papel ativo na doença. O principal fator ambiental associado à PAN é a infecção pelo HBV. Nesses pacientes, a doença manifesta‐se de maneira mais severa. Assim, a redução na prevalência dos casos de PAN nos últimos anos pode estar relacionada à diminuição nas taxas de infecção por HBV alcançadas por vacinação ampla da população, triagem de transfusões de sangue, tratamento mais eficaz da doença e a um melhor reconhecimento de outras vasculites necrosantes sistêmicas (p.ex., VAA, vasculite crioglobulinêmica).39,44
Febre, perda de peso, mialgia intensa e artralgia que afeta grandes articulações ocorrem em mais da metade dos pacientes.39 O espectro da doença varia desde o envolvimento de um único órgão à falência polivisceral, com uma marcante tendência a poupar os pulmões. Músculos e articulações, nervos periféricos e pele são os territórios mais frequentemente envolvidos, embora qualquer órgão possa ser acometido.43 Na maioria das vezes, o envolvimento neurológico manifesta‐se como mononeuropatia múltipla. No entanto, polineuropatia assimétrica e envolvimento do sistema nervoso central também podem ocorrer. As lesões cutâneas são vistas em 2%‐50% dos pacientes com PAN, apresentam‐se como púrpura ou urticária, livedo racemosa, úlceras grandes, nódulos subcutâneos e, mais raramente, infarto digital ou gangrena de extremidades.34,44
O envolvimento renal na PAN ocorre no nível das artérias pré‐glomerulares, causa hipertensão maligna e/ou insuficiência renal, mas sem glomerulonefrite, que é uma manifestação secundária com envolvimento de vasos menores. Os angiogramas abdominais e renais podem mostrar microaneurismas e/ou estenoses.34,44 A orquite, incluída nos critérios de classificação do ACR (tabela 3) e detectada em 24% dos pacientes, geralmente é unilateral e secundária à vasculite da artéria testicular.43 A dor abdominal ocorre em um terço dos pacientes. As manifestações digestivas podem ser severas, com risco de hemorragia e/ou perfuração, localizada principalmente no intestino delgado. As principais alterações oftalmológicas são descolamento bilateral de retina e vasculite retiniana.39
Histologicamente, as lesões inflamatórias vasculares são segmentares, predominam nos pontos de bifurcação. Na fase aguda, observa‐se necrose fibrinoide da camada média com um infiltrado composto inicialmente em sua maioria por neutrófilos polimorfonucleares, que se torna subsequentemente predominantemente linfo/histiocítico. Aneurismas e tromboses podem complicar lesões vasculares inflamatórias. A fase cicatricial é caracterizada por endarterite fibrosa, que pode levar à oclusão vascular. Pode‐se observar a coexistência de lesões em diferentes estágios em uma mesma biópsia. Não se observa inflamação granulomatosa.39 O ANCA, que está fortemente correlacionado com o envolvimento de pequenos vasos, é tipicamente negativo na PAN, embora Young et al.43 tenham mostrado 19% de positividade desses autoanticorpos em 27 pacientes coreanos que atendiam aos critérios do ACR 1990.
No momento do diagnóstico, um escore composto por cinco parâmetros (Five‐Factor Score – FFS) pode ser calculado para estimar o risco de mortalidade. Esses cinco fatores consistem em: proteinúria>1g/dia; insuficiência renal (creatinina sérica>1,58 mg/dL); sintomas gastrintestinais; miocardiopatia; e envolvimento do sistema nervoso central.43 A mortalidade entre os pacientes com um FFS de 2 é de 33,3%. O primeiro ano de doença é um período crítico para a ocorrência desse desfecho.33
Arterite cutâneaA arterite cutânea é uma vasculite necrosante crônica recorrente que afeta pequenas artérias e arteríolas na derme profunda e/ou hipoderme.35 Em uma revisão retrospectiva de 22 pacientes diagnosticados com essa vasculite, nosso grupo de estudo encontrou predominância em mulheres brancas, com média de 39,4 anos.45 Várias condições infecciosas (Streptococcus beta‐hemolítico do grupo A, HBV, HCV, HIV, parvovírus B19 e Mycobacterium tuberculosis) e não infecciosas (doenças autoimunes, doença inflamatória intestinal, neoplasias, imunização) foram relatadas em associação com a arterite cutânea. Medicamentos como penicilina e tetraciclina também estão relacionados à arterite cutânea e a minociclina é a principal envolvida. Nesses casos, observa‐se melhoria das lesões cutâneas com a retirada da medicação.40,46,47
A arterite cutânea está compreendida no grupo das vasculites mediadas por complexos imunes. A IFD geralmente revela depósitos de IgM e C3 na parede das artérias acometidas.44,45 Além disso, uma prevalência de 77,8% de anticorpos IgM contra o complexo fosfatidilserina‐protrombina em pacientes com arterite cutânea corrobora a teoria de que a protrombina ligada a células endoteliais apoptóticas induz uma resposta imune. Esse processo levaria, então, ao desenvolvimento de anticorpos do complexo antifosfatidilserina‐protrombina, que, por sua vez, ativam a via clássica do complemento, causam o dano vascular.44
As manifestações cutâneas mais frequentes são úlceras, livedo racemosa e nódulos dérmicos ou subcutâneos, localizados principalmente na porção inferior das pernas, região perimaleolar, que podem ascender até coxas e nádegas, ocasionalmente acometem mãos e pés. As úlceras regridem e deixam cicatrizes atróficas estelares, de cor marfínica, denominadas atrophie blanche (fig. 3), manifestação que pode ser observada em vasculopatia oclusivas, como a vasculopatia livedoide. A neuropatia periférica foi identificada em 25%‐32% dos pacientes com arterite cutânea.40,45 Embora menos comuns, os achados extracutâneos incluem ainda mal‐estar, febre, mialgia e artralgia.44,45

Morimoto e Chen identificaram nas amostras de biópsia de arterite cutânea quatro estágios: agudo, subagudo, reparativo e cicatricial, de acordo com o tipo de infiltrado inflamatório predominante e as alterações morfológicas observadas na parede vascular. Eles variam desde infiltração proeminente de neutrófilos na parede dos vasos à inflamação celular mínima com oclusão do lúmen vascular por trombos de fibrina e neovascularização. Múltiplos estágios podem coexistir na mesma ou em diferentes amostras de biópsia de um paciente.48
Nakamura et al.40 estabeleceram critérios clínicos e histológicos que devem estar presentes para confirmar o diagnóstico da doença. Eles estão representados na tabela 4. Uma investigação detalhada sobre condições autoimunes, trombofilias e doenças infecciosas deve ser feita. Em países onde a tuberculose é endêmica, o teste de Mantoux é particularmente importante.45
Critérios diagnósticos para arterite cutânea, anteriormente denominada poliarterite nodosa cutânea
| Achados clínicos compatíveis | Nódulos subcutâneos, livedo, púrpura ou úlceras. |
| Achados histopatológicos | Vasculite necrotizante fibrinoide das artérias de pequeno e médio calibre. |
| Manifestações de exclusão | Febre (≥ 38° C, ≥ duas semanas) |
| Perda de peso (≥ 6 kg em seis meses); | |
| Hipertensão; | |
| Insuficiência renal rapidamente progressiva, infarto renal; | |
| Hemorragia cerebral, infarto cerebral; | |
| Infarto do miocárdio, doença cardíaca isquêmica, pericardite, insuficiência cardíaca; | |
| Pleurite; | |
| Hemorragia intestinal, infarto intestinal; | |
| Neuropatia periférica fora da região de pele afetada; | |
| Artralgia/artrite ou mialgia/miosite fora da região de pele afetada; | |
| Arteriografia anormal (múltiplos microaneurismas, estenose e obliteração). |
A arterite macular linfocítica (AML), também conhecida como arterite trombofílica linfocítica, apresenta‐se clinicamente como máculas ou placas hiperpigmentadas e livedo racemosa, preferencialmente nos membros inferiores. Embora AML seja mais comumente assintomática, pode haver uma propensão ao envolvimento neurológico semelhante a outras vasculites cutâneas. A patogênese dessa entidade ainda não está clara e persiste o debate se a AML representa uma nova síndrome de vasculite cutânea ou uma variante indolente de arterite cutânea. Histologicamente, é caracterizada por um denso infiltrado de células mononucleares na parede muscular das arteríolas da junção dermo‐subcutânea, com estreitamento variável de seu lúmen por um típico anel hialinizado de fibrina (fig. 4). Ao contrário da descrição original, durante o exame histopatológico, ruptura da lâmina elástica interna foi detectada em alguns pacientes.49,50 A depender do momento de feitura da biópsia e da seção examinada, os estágios subagudo e reparativo da arterite cutânea podem ser confundidos com a AML. Por vezes, biópsias profundas, repetidas, e análise de cortes seriados são necessárias antes de concluir que não há arterite necrosante, uma vez que o envolvimento do vaso é segmentar e focal.42,48
Deficiência de adenosina desaminase tipo 2 (ADA2) mostra lesões tipo livedo sem ulceração nas pernas e respectivo exame histopatológico demonstra na junção dermo‐hipodérmica um vaso arterial circundado por infiltrado linfocitário e com anel de fibrina na camada íntima.")
Descobertas recentes atribuíram a uma mutação autossômica recessiva no gene CERC1 no cromossomo 22q11 um dos defeitos genéticos associados à arterite cutânea. O gene CERC1 codifica para a adenosina desaminase tipo 2 (ADA2), um grupo de enzimas envolvidas no metabolismo das purinas. A ADA2 é sintetizada apenas pelo sistema monocítico‐macrofágico, células com importante função na regulação da resposta imune. Várias mutações que podem ser homozigotas ou heterozigotas compostas têm sido detectadas, embora sua real prevalência em todo o mundo permaneça desconhecida. A história familiar é negativa na maioria dos casos e a idade de início do quadro varia de 2 meses a 59 anos.51,52
A deficiência de ADA2 é uma síndrome autoinflamatória que cursa com diversas anormalidades imunes, como aumento de macrófagos pró‐inflamatórios M1 (em oposição a macrófagos anti‐inflamatórios M2), compromete a integridade endotelial e estabelece um círculo vicioso de vasculopatia e inflamação. Uma proporção significativa de crianças desenvolve características de imunodeficiência de células B, inclusive infecções recorrentes, baixos níveis de Ig e citopenia que envolvem uma ou mais linhagens. As manifestações clínicas variam em gravidade, desde envolvimento cutâneo limitado a vasculite multissistêmica grave com febre recorrente, hipertensão renovascular/aneurismas da artéria renal, hepatoesplenomegalia com hipertensão portal, neuropatia periférica e acidente vascular cerebral antes dos 5 anos. Os achados cutâneos incluem: livedo racemosa, nódulos subcutâneos, ulcerações, fenômeno de Raynaud e necrose de dígitos nos casos mais severos. Nas biópsias de pele, em geral, observa‐se vasculite necrotizante não granulomatosa de artérias de médio calibre geralmente; no entanto, alguns pacientes podem apresentar vasculite leucocitoclástica ou mesmo presença de linfócitos T perivasculares, sem franca vasculite.51–54
Vasculite nodularA vasculite nodular (VN) consiste em uma paniculite predominantemente lobular, que se apresenta com algum tipo de vasculite na maioria dos casos. A idade dos indivíduos acometidos varia de 23 a 81 anos (média de 55,8 anos).55 As pacientes típicas são mulheres obesas com algum grau de insuficiência venosa, que apresentam episódios de nódulos e placas na região posterolateral dos membros inferiores durante os meses de inverno (fig. 5). Podem ainda ocorrer lesões nas coxas, glúteos e antebraços. Essas áreas são usualmente sensíveis ou dolorosas apenas à pressão local e frequentemente evoluem com ulceração focal e drenagem, causam cicatrizes e hiperpigmentação pós‐inflamatória. A doença costuma seguir um curso crônico, com episódios de agudização a cada três ou quatro meses. Os pacientes apresentam boa saúde geral e não manifestam sinais e sintomas sistêmicos.55–57

Por anos, a VN esteve associada unicamente à infecção por Mycobacterium tuberculosis, como manifestação tipo tubercúlide. No entanto, tem sido descrita em pacientes com outras condições infecciosas, como HBV, nocardia, pseudomonas e fusarium; doenças não infecciosas, como hipotireoidismo, leucemia linfocítica crônica, artrite reumatoide e doença de Crohn; e possivelmente também como um quadro induzido por medicações, uma vez que já foi descrita durante o tratamento de psoríase com etarnecepte, um inibidor do fator de necrose tumoral (TNF)‐α e em associação com o uso de propiltiouracil, com rápida resolução após descontinuação.55
O padrão histopatológico mais comumente encontrado é o de vasculite que envolve pequenas vênulas localizadas no centro do lóbulo adiposo. Veias e, menos frequentemente, artérias septais podem estar acometidas simultânea ou isoladamente. Em alguns casos, observa‐se apenas endarterite obliterante. Pode haver necrose extensa dos adipócitos dos lóbulos do tecido subcutâneo. Infiltrado misto de histiócitos epitelioides, células gigantes multinucleadas e linfócitos contribuem para o achado de uma paniculite granulomatosa lobular nas lesões completamente desenvolvidas. As colorações especiais não demonstram a presença de bacilos ácido‐álcool resistentes. A investigação de pacientes com suspeita de VN deve incluir a pesquisa de fragmentos do DNA do M. tuberculosis em amostras teciduais pela técnica de PCR, radiografia de tórax, teste tuberculínico e sorologias para HBV e HCV. Raramente detecta‐se evidência de tuberculose sistêmica ativa.55,56
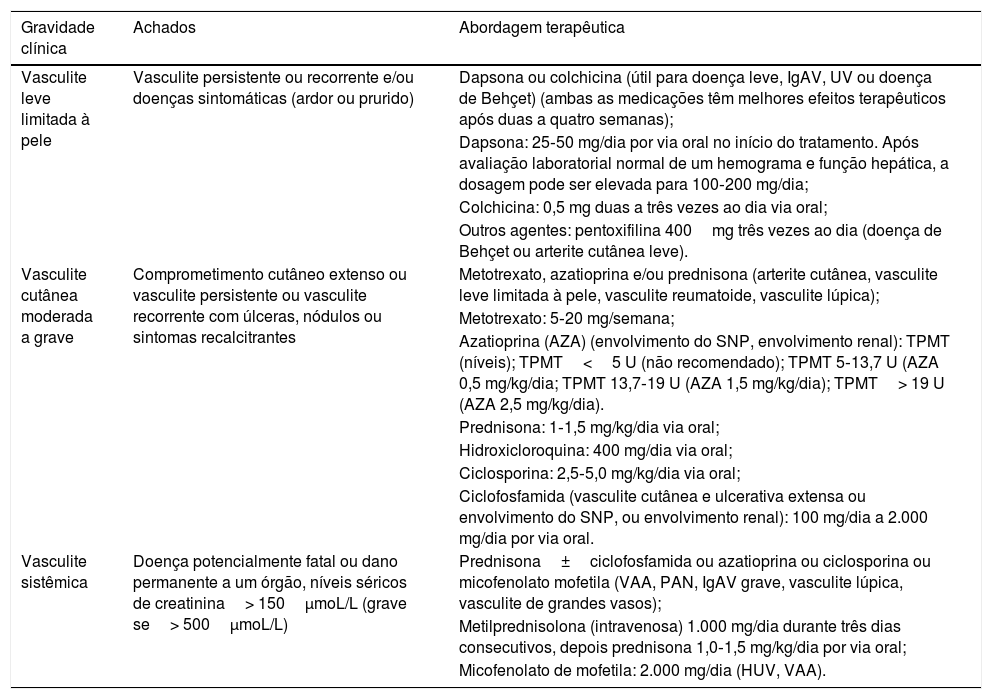
Tratamento das vasculitesVasculites cutâneasNa maioria dos casos, as vasculites cutâneas apresentam um curso autolimitado. Uma vez excluído o envolvimento sistêmico, os procedimentos terapêuticos devem ser concentrados no tratamento dos sintomas, na prevenção de gatilhos (como medicamentos e exercícios prolongados) e no tratamento de infecções subjacentes. Repouso, elevação dos membros inferiores e uso de meias de compressão podem ser úteis para diminuir a deposição de complexos imunes relacionados à estase e acelerar a cicatrização das úlceras. Não há evidências disponíveis sobre o benefício de corticosteroides tópicos nas VCPV. Seu único efeito parece ser a redução da resposta inflamatória local, que alivia o prurido e a sensação de queimação. Muitas vezes, medicamentos anti‐inflamatórios não esteroides, principalmente ácido acetilsalicílico (1 a 3 g/d) e indometacina (25 a 50 mg/três vezes ao dia), são suficientes, sem que haja necessidade de se associarem corticosteroides sistêmicos.4,58,59
As opções terapêuticas para uso prolongado incluem a colchicina, particularmente útil para sintomas de pele e articulações (0,5 mg/duas a três vezes ao dia, dose limitada pelos efeitos colaterais gastrintestinais). A colchicina diminui a degranulação neutrofílica e modifica a expressão de moléculas de adesão endotelial. A dapsona, cujos efeitos anti‐inflamatórios ocorrem por meio da inibição da migração de neutrófilos para áreas de lesão tecidual, bem como inibição da atividade da mieloperoxidase e de enzimas lisossomais dos neutrófilos, tem eficácia comprovada quando usada isoladamente ou em associação com a colchicina (dose inicial 25‐50 mg, titulada até 200 mg/dia). A dapsona é particularmente útil em pacientes com EED. Sua prescrição exige uma triagem para deficiência de glicose‐6‐fosfato desidrogenase e monitoração laboratorial regular para anemia hemolítica, metemoglobinemia e, menos comumente, agranulocitose. Outros tratamentos para as VCPV da pele incluem hidroxicloroquina (200‐400 mg/dia), particularmente benéfica na UV e na prevenção de crises de vasculite em pacientes com LES inativo; e pentoxifilina (400‐1.200 mg/dia), cujos benefícios da associação com a dapsona superam os resultados obtidos com o uso de quaisquer uma dessas duas drogas empregadas isoladamente. Os anti‐histamínicos H1 (hidroxizina 25 mg/dia) isoladamente ou em combinação com os anti‐histamínicos H2 (ranitidina 150 mg/duas vezes por dia) podem ser usados para aliviar o prurido, ao bloquear a liberação de histamina, além de diminuir a permeabilidade vascular aos imunocomplexos.4,21,58
Quando nenhum desses agentes é eficaz ou tolerado, e as VCPV da pele são significativamente sintomáticas, e/ou extensamente ulcerativas, corticoides sistêmicos tornam‐se a principal opção terapêutica (0,5 a 1 mg/kg/dia de prednisona ou prednisolona). Para os casos persistentes/resistentes, a monoterapia com prednisona não é recomendada devido à possibilidade de efeitos adversos, sobretudo em crianças, prefere‐se então seu uso em baixas doses como adjuvante do tratamento com agentes poupadores de corticosteroides, como azatioprina, metotrexato e micofenolato de mofetila. Ciclofosfamida e ciclosporina podem ser eficazes. Outros tratamentos específicos, como o da crioglobulinemia, podem incluir plasmaférese para remoção de complexos imunes.58–60
Vasculites sistêmicasA terapia padrão para as vasculites sistêmicas é iniciada com altas doses de corticosteroides orais (1‐1,5 mg/kg/dia até 80 mg). Quando a remissão é alcançada, a diminuição precisa ser feita de maneira lenta e progressiva, a fim de se obter uma dose diária de prednisona na faixa de 20 mg/dia aos três meses, 10 mg/dia aos seis meses e 5 mg/dia aos 12 meses até sua retirada entre 12 e 24 meses. Assim, como nas VCPV exclusivas da pele, nas formas graves ou recorrentes, os corticosteroides devem sempre estar associados à terapia imunossupressora. A ciclofosfamida pode ser administrada por via oral (500 mg/d a 2 g/kg/dia) ou intravenosa (600 mg/m2, a cada 2‐4 semanas), essa geralmente preferida por apresentar eficácia comparável com menor taxa de efeitos colaterais. Uma vez alcançada a remissão (três a seis meses), a mudança para um regime de manutenção com metotrexato (15 a 25 mg/semana), azatioprina (2 mg/kg/dia) ou ciclosporina (2,5‐5,0 mg/kg/dia administrados por via oral, divididos em duas doses) é recomendada, a fim de se evitar as possíveis complicações da terapia com ciclofosfamida. O metotrexato oral ou parenteral pode ser usado como uma opção menos tóxica à ciclofosfamida para a indução de remissão nas VAA sem risco à vida ou de lesão de órgão‐alvo. Micofenolato de mofetila (2 g/dia por via oral) ou leflunomida são usados em pacientes intolerantes ou não responsivos ao metotrexato ou à azatioprina.3,7,30
As opções de tratamento para os casos refratários incluem o anticorpo monoclonal anti‐CD20 rituximabe (500 mg a cada seis meses por 18 meses) e os inibidores do TNF‐α (infliximabe ou etanercepte), bem como a imunoglobulina intravenosa (200 a 1.000mg/kg/dia). Os inibidores de TNF‐α são úteis na indução de remissão da deficiência de ADA2, enquanto outros imunossupressores não têm alcançado os resultados esperados. A terapia de manutenção para as vasculites sistêmicas deve continuar por 18 a 24 meses após a remissão, devido à elevada frequência de recidivas. Nos pacientes com PAN associada ao HBV, os regimes convencionais de tratamento sem terapia antiviral são contraindicados devido ao risco de viremia contínua, progressão da hepatite crônica ou cirrose e, mais ameaçadoramente, reativação do vírus com hepatite fulminante. Assim, recomenda‐se uma combinação de corticosteroides, plasmaférese e terapia antiviral. A plasmaférese é empregada também para pacientes com doença renal grave rapidamente progressiva, a fim de melhorar a sobrevida; nesse cenário, ela é comprovadamente superior aos pulsos de metilprednisolona (1 g/dia por três dias). O tratamento das vasculites aqui abordadas está resumido na tabela 5. A posologia dos medicamentos listados considera a dose para pacientes adultos.
Abordagem terapêutica para vasculite de acordo com a gravidade das manifestações
| Gravidade clínica | Achados | Abordagem terapêutica |
|---|---|---|
| Vasculite leve limitada à pele | Vasculite persistente ou recorrente e/ou doenças sintomáticas (ardor ou prurido) | Dapsona ou colchicina (útil para doença leve, IgAV, UV ou doença de Behçet) (ambas as medicações têm melhores efeitos terapêuticos após duas a quatro semanas); |
| Dapsona: 25‐50 mg/dia por via oral no início do tratamento. Após avaliação laboratorial normal de um hemograma e função hepática, a dosagem pode ser elevada para 100‐200 mg/dia; | ||
| Colchicina: 0,5 mg duas a três vezes ao dia via oral; | ||
| Outros agentes: pentoxifilina 400mg três vezes ao dia (doença de Behçet ou arterite cutânea leve). | ||
| Vasculite cutânea moderada a grave | Comprometimento cutâneo extenso ou vasculite persistente ou vasculite recorrente com úlceras, nódulos ou sintomas recalcitrantes | Metotrexato, azatioprina e/ou prednisona (arterite cutânea, vasculite leve limitada à pele, vasculite reumatoide, vasculite lúpica); |
| Metotrexato: 5‐20 mg/semana; | ||
| Azatioprina (AZA) (envolvimento do SNP, envolvimento renal): TPMT (níveis); TPMT<5 U (não recomendado); TPMT 5‐13,7 U (AZA 0,5 mg/kg/dia; TPMT 13,7‐19 U (AZA 1,5 mg/kg/dia); TPMT> 19 U (AZA 2,5 mg/kg/dia). | ||
| Prednisona: 1‐1,5 mg/kg/dia via oral; | ||
| Hidroxicloroquina: 400 mg/dia via oral; | ||
| Ciclosporina: 2,5‐5,0 mg/kg/dia via oral; | ||
| Ciclofosfamida (vasculite cutânea e ulcerativa extensa ou envolvimento do SNP, ou envolvimento renal): 100 mg/dia a 2.000 mg/dia por via oral. | ||
| Vasculite sistêmica | Doença potencialmente fatal ou dano permanente a um órgão, níveis séricos de creatinina> 150μmoL/L (grave se> 500μmoL/L) | Prednisona±ciclofosfamida ou azatioprina ou ciclosporina ou micofenolato mofetila (VAA, PAN, IgAV grave, vasculite lúpica, vasculite de grandes vasos); |
| Metilprednisolona (intravenosa) 1.000 mg/dia durante três dias consecutivos, depois prednisona 1,0‐1,5 mg/kg/dia por via oral; | ||
| Micofenolato de mofetila: 2.000 mg/dia (HUV, VAA). |
IgAV, vasculite por IgA; HUV, urticária vasculite hipocomplementêmica; PAN, poliarterite nodosa; TPMT, tiopurina S‐metiltransferase; VAA, vasculites associadas ao ANCA.
O diagnóstico e o manejo das vasculites devem ser multidisciplinares na maioria dos casos. Na prática clínica, uma investigação acerca do envolvimento de órgãos e sistemas internos é obrigatória como primeiro passo diante de pacientes com lesões cutâneas sugestivas de vasculite.
Suporte financeiroCoordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), Código de Financiamento 00.
Contribuição dos autoresThâmara Cristiane Alves Batista Morita: Elaboração e redação do manuscrito.
Paulo Ricardo Criado: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Roberta Fachini Jardim Criado: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; revisão crítica do manuscrito.
Gabriela Franco S. Trés: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Mirian Nacagami Sotto: Aprovação da versão final do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Como citar este artigo: Morita TCAB, Criado PR, Criado RFJ, Trés GFS, Sotto MN. Update on vasculitis: overview and relevant dermatological aspects for the clinical and histopathological diagnosis – Part II. An Bras Dermatol. 2020;95:493–507.
Trabalho realizado no Departamento de Dermatologia, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil.
