

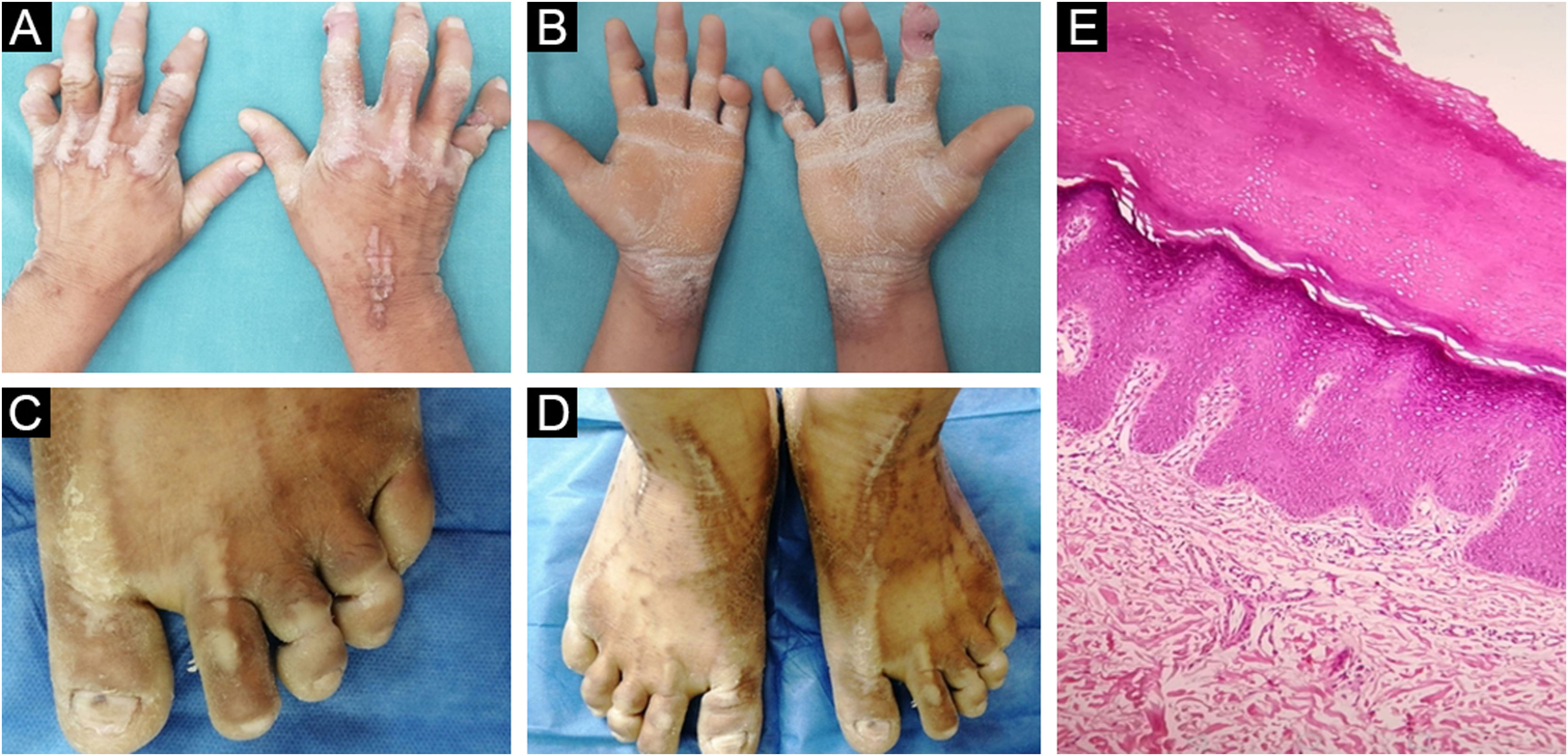
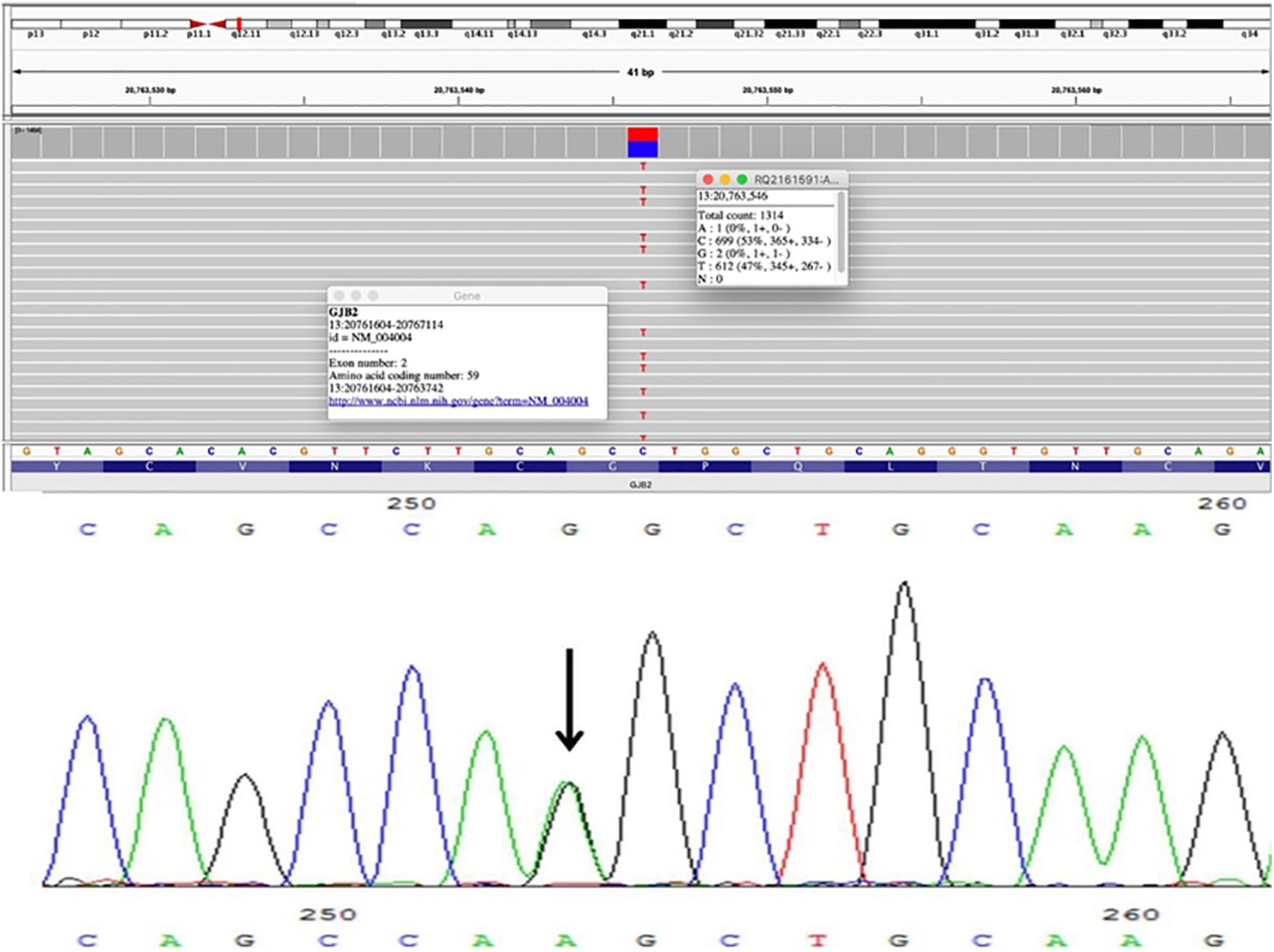
Um pescador de 31 anos compareceu à clínica dermatológica em virtude de uma úlcera com evolução de três meses no quinto dedo da mão direita. O paciente apresentava história de perda auditiva bilateral grave desde a infância e faixas constritivas nas falanges distais das mãos e dos pés. Essas bandas haviam sido liberadas há cinco anos através de cirurgia plástica com vários enxertos de pele. O exame físico revelou hiperceratose palmoplantar com aspecto de favo de mel e lesões ceratóticas bilaterais lineares e em formato de estrela‐do‐mar no dorso dos pés e nas articulações metacarpofalângicas (fig. 1). Foram observadas também placas ceratóticas nos cotovelos e joelhos. Não havia história familiar de lesões cutâneas semelhantes ou perda auditiva. Uma amostra de DNA genômico foi obtida de leucócitos do sangue periférico. A análise histopatológica da placa do cotovelo revelou hiperceratose ortoceratótica (fig. 1). Foram realizados testes genéticos moleculares por meio de sequenciamento e análise de deleção/duplicação de 203 genes relacionados a um painel abrangente para surdez, com identificação de variante patogênica no éxon 2 do gene GJB2, c.175G>A (p. Gly59Ser) em estado de heterozigose (fig. 2). O diagnóstico de síndrome de Vohwinkel (SV) foi concluído e iniciado tratamento conservador com emolientes e terapia ceratolítica tópica.
 Lesões ceratóticas lineares bilaterais no dorso das articulações metacarpofalângicas e punho direito. (B) Ceratodermia palmar bilateral com aspecto de “favo de mel”. (C) Pseudoainhum no terceiro e quarto artelhos. (D) Lesões ceratóticas bilaterais em forma de estrela‐do‐mar no dorso dos pés. E, Hiperceratose ortoceratótica (Hematoxilina & eosina, 100×.")
Aspecto clínico e histopatológico. (A) Lesões ceratóticas lineares bilaterais no dorso das articulações metacarpofalângicas e punho direito. (B) Ceratodermia palmar bilateral com aspecto de “favo de mel”. (C) Pseudoainhum no terceiro e quarto artelhos. (D) Lesões ceratóticas bilaterais em forma de estrela‐do‐mar no dorso dos pés. E, Hiperceratose ortoceratótica (Hematoxilina & eosina, 100×.
 no éxon 2.")
A SV (OMIM #124500) é ceratodermia palmoplantar genética rara com herança autossômica dominante que se manifesta em bebês e se torna evidente na idade adulta.1,2 Sua prevalência e incidência são desconhecidas em decorrência do pequeno número de casos publicados. No entanto, tem sido relatada com frequência em mulheres brancas.2,3 A SV está associado a perda auditiva neurossensorial em virtude de variantes patogênicas na sequência codificadora do gene GJB2 (éxon 2, localizado no cromossomo 13q12.11) que codifica a conexina 26, uma proteína de junção beta‐2 composta por 226 aminoácidos.1 As conexinas agregam‐se em grupos de seis em torno de um poro central de 2 a 3nm para formar um conéxon. Conéxons de células adjacentes ligam‐se de maneira covalente formando um canal entre as células. Grandes coleções de conéxons, chamadas placas, são os constituintes das junções gap ou comunicantes. As junções comunicantes tornam possível a troca intercelular direta de íons e moléculas através de seus poros aquosos centrais, possibilitam a sincronização da atividade em tecidos excitáveis e a troca de metabólitos e moléculas sinalizadoras em tecidos não excitáveis. As junções comunicantes são expressas em queratinócitos humanos, cóclea, folículos capilares e unhas, aumentando a sobrevivência dos queratinócitos epidérmicos e a diferenciação terminal.4
Essa alteração de sequência c.175G>A (p. Gly59Ser) detectada no paciente do presente caso resulta em substituição de aminoácidos da glicina pela serina no códon 59da proteína GJB2. O resíduo de glicina é altamente conservado e há uma pequena diferença físico‐química entre a glicina e a serina, representando efeito deletério de perda de função na proteína. Em virtude dessa substituição, pode ocorrer uma alteração na volta reversa da primeira alça extracelular da conexina 26, que está associada ao controle de voltagem e à interação intercelular.5,6 A modelagem avançada da sequência proteica e propriedades biofísicas (ou seja, estruturais, funcionais e espaciais, conservação de aminoácidos, variação físico‐química, mobilidade de resíduos e estabilidade termodinâmica) realizadas na Invitae indicam que se espera que essa variante missense interrompa a função da proteína GJB2. Bondenson et al.5 relataram um caso semelhante sem história familiar de SV, portanto pode‐se concluir que essa variante é uma nova mutação.
O paciente do presente caso negou qualquer dermatose visível em familiares de primeiro grau, mas por questões econômicas não foi possível testar nenhum outro membro da família. Ao exame físico, os pacientes com SV apresentam perda auditiva associada à hiperceratose palmoplantar com aspecto típico de favo de mel, bandas de constrição que levam ao estrangulamento e autoamputação nas articulações interfalângicas das mãos e pés (pseudoainhum) e ceratoses lineares e em forma de estrela‐do‐mar nos cotovelos, joelhos e dorso das mãos e pés.7,8 A histopatologia em geral é inespecífica, e apenas hiperceratose com ortoceratose e paraceratose são frequentemente observadas. O diagnóstico diferencial considera pacientes que apresentam perda auditiva associada a doenças cutâneas. Isso inclui a síndrome de Bart‐Pumphrey, ceratodermia palmoplantar, síndrome de ceratite‐ictiose‐surdez, ceratodermia acral, ceratodermia palmoplantar de Sybert, doença de Meleda e síndrome de ictiose‐surdez tipo hystrix.3 Até o momento, não há tratamento específico para SV. A ceratodermia e as constrições são tratáveis com retinoides tópicos e orais, bem como com cirurgia, com resultados variáveis.9 Entretanto, na maioria dos pacientes com SV, a expectativa de vida não é prejudicada. No presente caso, foi impossível iniciar retinoides orais por razões econômicas; entretanto, foram iniciados monitorização dermatológica rigorosa das constrições e uso de emolientes.
Suporte financeiroNenhum.
Contribuição dos autoresMaría Caridad Duran‐Lemarie: Elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; revisão crítica da literatura; aprovação da versão final do manuscrito.
Luis Enrique Cano‐Aguilar: Elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Edmar Obed Benitez‐Alonso: Revisão crítica da literatura; obtenção, análise e interpretação dos dados.
Dalia Cruz‐Sotomayor: Revisão crítica da literatura.
Ulises Villela‐Segura: Revisão crítica da literatura.
Hector Proy‐Trujillo: Participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Conflito de interessesNenhum.
Os autores agradecem à Invitae Corporation (1400 16th Street, São Francisco, CA 94103, #05D2040778) pelo suporte técnico contínuo, análise de dados e interpretação dos resultados genômicos.
Como citar este artigo: Duran‐Lumarie MC, Cano‐Aguilar LE, Benitez‐Alonso EO, Cruz‐Sotomayor D, Villela‐Segura U, Proy‐Trujillo H. Vohwinkel syndrome with De novo mutation in the GJB2 gene with Heterozygous mutation c.175G>A (p. Gly59Ser). An Bras Dermatol. 2025;100. https://doi.org/10.1016/j.abd.2023.01.010.
Trabalho realizado no Dermatology Center of Yucatán, Mérida, México.
