











A psoríase pustulosa generalizada (von Zumbusch) é rara erupção aguda caracterizada por múltiplas pústulas estéreis sobre base eritematoedematosa eventualmente associada à psoríase em placas. Classicamente, manifesta quadro sistêmico potencialmente grave e demanda diagnóstico e intervenção precoces. A duração do surto e o intervalo entre os episódios pustulosos é extremamente variável. Recentemente, alterações genéticas têm sido identificadas principalmente nas variantes familiares e precoces da doença. O arsenal terapêutico para a psoríase pustulosa generalizada é limitado; no entanto, novos medicamentos em estudo almejam o controle não somente da erupção mas também das recidivas da doença.
Quadros inflamatórios que cursam com pústulas estéreis sobre base eritematosa, de topografia e extensão variável e que refletem a infiltração de neutrófilos na epiderme são característicos da forma pustulosa da psoríase. Essa variante pode advir de ou coexistir com lesões crônicas de psoríase em placas ou surgir e recorrer na ausência de qualquer outra forma de psoríase. É a causa mais comum de pustulose cutânea.1
Existem quatro subtipos de psoríase pustulosa: 1) psoríase pustulosa generalizada von Zumbusch (PPG), 2) psoríase pustulosa anular ou circinada, 3) psoríase pustulosa exantemática e 4) psoríase pustulosa localizada, incluindo as variantes de psoríase pustulosa palmoplantar (PPP) e acrodermatite contínua de Hallopeau (ACH). Variantes mistas também são descritas.1
A PPG é variante aguda e multissistêmica grave da psoríase e se manifesta por meio de placas eritematoedematosas sobre as quais surgem múltiplas pústulas, associadas ou não à forma em placas. Nesta revisão, serão abordados os principais aspectos da PPG em relação à epidemiologia, genética, fisiopatologia, quadro clínico, terapêutica e prognóstico dessa rara entidade.
EpidemiologiaDesde o relato original por Leo Ritter von Zumbusch (Viena, 1874‐1940) em 1909,2 que descreveu a ocorrência de PPG em irmãos bem como a evolução da moléstia, discute‐se a classificação da PPG como variante mais grave da psoríase em placas ou entidade clínica distinta.1
A prevalência estimada da PPG é maior em populações asiáticas (7,46 por milhão no Japão)3 do que em caucasianos (1,76 por milhão na França).4 Ocorre com maior prevalência no sexo feminino e pode surgir em qualquer faixa etária, mas é mais frequente em adultos entre a quarta e a quinta décadas de vida.3–5 Dados de registro com 15.794 indivíduos com psoríase de população asiática na Malásia estimam a prevalência da PPG em torno de 1%,6 enquanto estudo epidemiológico no Japão relatou a ocorrência em 1,3% de um total de 11.631 casos de psoríase.7 A associação com psoríase em placas tem sido reportada com variação entre 25 a 30% até 65% dos casos.8,9 Na população pediátrica, parece ocorrer com maior freqüência no sexo masculino (3:2), com variação de 0,6 a 7% dos casos de psoríase.5,10
Atualmente, considera‐se a PPG essencialmente uma entidade distinta da psoríase em placas, de perfil genético e patogênico distintos.5,11 Hussain et al. observaram que nos pacientes com mutação no gene do receptor do antagonista da interleucina‐36 (IL‐36RN), o aparecimento da PPG tende a ser mais precoce, associado a maior risco de inflamação sistêmica e a menor associação com psoríase em placas.12
GenéticaA base genética da psoríase em placas é evidente pela maior prevalência da doença entre parentes de primeiro grau, duas a três vezes maior do que na população geral. Muitos estudos de varredura genômica reproduzidos em diferentes populações demonstraram polimorfismos genéticos associados à psoríase em placas.13 As regiões cromossômicas ou loci de suscetibilidade à psoríase são mais de 50, o que demonstra a característica de interação multigênica e epigenética da doença.14 Os genes cujos polimorfismos conferem suscetibilidade à psoríase em placa são relacionados à apresentação de antígenos (ERAP1 e MHC), sinalização da via interferon (IL‐28RA e IFIH1), da via NFkB (ZNF313, REL, TNIP1, TNFAIP3, NFkB1A),15 da IL‐17 (TRAF3IP2), da IL‐23 (TYK2, IL‐23R, IL‐23A, IL‐12B) e à barreira epidérmica (LCE3D).16
Responsável por mais de 50% da herdabilidade da doença, os genes do complexo principal de histocompatibilidade (MHC) que codificam antígenos leucocitários de membrana (HLA) estão localizados num segmento genômico na posição 6p.21, com cerca de 15 outros genes. O alelo de suscetibilidade mais relacionado à psoríase é o HLA‐Cw*0602. As moléculas do complexo de histocompatibilidade de classe I (HLA) reconhecem antígenos não próprios e os apresenta aos linfócitos T CD8+para iniciar resposta imune e favorecer a sinalização entre diferentes tipos celulares.16
Já os estudos genéticos das várias formas de psoríase pustulosa revelam diferenças com a psoríase em placas. Polimorfismos HLA e genes da barreira epidérmica não conferem suscetibilidade a essa forma da doença. Entretanto, mutações em genes relacionados à imunidade inata, como IL‐36RN, AP1S3 e CARD14, são importantes em parte dos pacientes.17
A análise de 5.249 pacientes com diferentes formas de psoríase pustulosa mostrou que as mutações no IL36RN são a anormalidade genética mais frequentemente observada na psoríase pustulosa.18 Os genes codificadores da família da IL‐36 estão localizados no cromossomo 2q13.18
Mutações autossômicas recessivas da perda de função no gene IL‐36RN são encontradas em aproximadamente 25% dos casos de PPG. Esse gene codifica o antagonista do receptor da IL‐36 (IL‐36Ra), que modula a atividade das citocinas da família IL‐1 (IL‐36α, ‐β e ‐γ). As análises genótipo‐fenótipo indicam que os alelos mutados são menos comuns em indivíduos com PPP do que PPG e ACH e, quando há uma única mutação no gene IL‐36RN, a doença tende a se expressar mais tardiamente. Ocorre penetrância variável dos alelos e modificadores genéticos, e fatores ambientais também influenciam sua expressão.19 Mutações homozigóticas no gene IL‐36RN estão associadas a idade mais precoce de início em todas as variantes da psoríase pustulosa, evolução mais grave e resposta terapêutica diferente da psoríase em placas.18
O gene AP1S3 (adaptor‐related protein complex 1, sigma‐3 subunit) está localizado na posição cromossômica 2q36.1. Os complexos de proteínas adaptadoras (AP) são heterotetrâmeros citosólicos que promovem a produção e a mobilização de pequenas vesículas transportadoras. O AP‐1 é dedicado ao transporte entre a rede trans‐Golgi e os endossomos. A subunidade sigma 3 é a principal estabilizadora da AP‐1 e é codificada pelo gene AP1S3. A AP‐1 é a responsável pela formação de autofagossomos, estruturas intracelulares para degradação de proteínas e organelas danificadas. Autofagia defeituosa leva a acúmulo de p62 (proteína adaptadora na ativação de NF‐kB) e regulação positiva da inflamação por IL‐36.19 Mutações no gene AP1S3 foram encontradas em 7 a 12% de pacientes europeus. No estudo de Twelves et al., as mutações no AP1S3 foram encontradas com frequência comparável entre os tipos de doenças. As mutações conhecidas são p.Phe4Cys e p.Arg33Trp.18
O gene CARD14 (caspase recruitment domain‐containing protein 14, também chamado de CARD‐containing MAGUK protein 2 ou Carma 2) localiza‐se na posição cromossômica 17q25.3. É altamente expresso em queratinócitos e codifica uma proteína que, após oligomerização, intermedia a ativação por TRAF‐2da sinalização de NF‐κB; é o ordenador dessa ativação e das vias de sinalização MAPK por meio do recrutamento MALT e BCL10. Leva à estimulação de genes pro‐inflamatórios IL‐36, IL‐8, CCl20 e peptídios antimicrobianos.20,21 Uma substituição de ganho de função no CARD14 foi associada à PPG. Também foram descritas em psoríase em placas e PPP, indicando mecanismos compartilhados. O CARD14 expresso em queratinócitos associa‐se ao complexo de sinalização ACT1‐TRAF6 e faz intermediação da ativação da via de sinalização de NF‐κB e MAPK induzida por IL‐17A, o que leva à expressão de fatores pró‐inflamatórios. Assim, é também mediador‐chave de sinalização da IL‐17A.18
No estudo de Twelves et al., as mutações do CARD14 foram observadas em apenas oito pacientes. O único alelo da doença associado à psoríase pustulosa (p.Asp176His) ou em placas (p.Gly117Ser) foi encontrado em pacientes com PPG de descendência chinesa, e não foram detectados entre pacientes europeus.18
Este estudo também teve o objetivo de avaliar as características clínicas e genéticas da psoríase pustulosa em uma coorte de pacientes não relacionados (251 com PPG, 560 com PPP, 28 com APH e 24 com múltiplos diagnósticos). Os alelos mutados IL‐36RN estavam presentes em uma variedade de grupos étnicos, com maior prevalência observada entre pacientes de descendência europeia (34,7%) e asiática (28,8%), e foi maior em PPG e APH (23,7% e 18,2%, respectivamente) em comparação com PPP (5,2%). Uma análise da variante recorrente p.Ser113Leu mostrou que sua frequência em pacientes britânicos era quase 10 vezes maior do que a observada na população controle.18 Essa variante também foi relatada em dois casos de pacientes brasileiros.22 Os alelos de AP1S3 tiveram frequência semelhante e baixa em todos os subtipos da doença. Alelos de risco em dois loci distintos (IL‐36RN e AP1S3; IL‐36RN e CARD14) foram relatados em vários casos, mostrando que a heterogeneidade genética é complexa, e que pleiotropia e herança digênica podem influenciar a expressão da doença.18
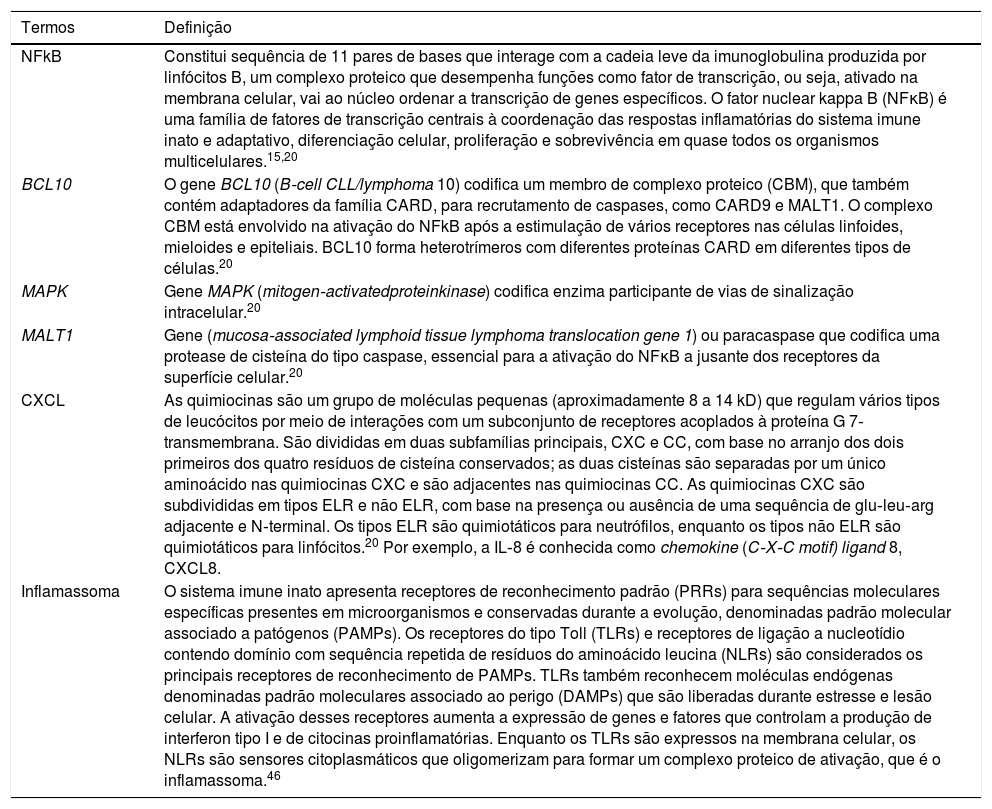
ImunologiaO transcriptoma da PPG é altamente associado a genes do sistema imune inato, embora compartilhe características da psoríase em placas (tabela 1).18,23 Os níveis de expressão de IL‐1β, IL‐1RN, IL‐36α, IL‐36β, IL‐36γ e IL‐36Ra aumentam na PPG e na psoríase em placas, em comparação com a pele normal, e são mais intensos na forma pustulosa.18,23 A sinalização maior para IL‐1 e IL‐36 na PPG está relacionada ao aumento significativo de expressão de substâncias quimiotáticas para neutrófilos (CXCL1, CXCL2 e CXCL8).23
Glossário explicativo de termos envolvidos na imunologia da psoríase pustulosa
| Termos | Definição |
|---|---|
| NFkB | Constitui sequência de 11 pares de bases que interage com a cadeia leve da imunoglobulina produzida por linfócitos B, um complexo proteico que desempenha funções como fator de transcrição, ou seja, ativado na membrana celular, vai ao núcleo ordenar a transcrição de genes específicos. O fator nuclear kappa B (NFκB) é uma família de fatores de transcrição centrais à coordenação das respostas inflamatórias do sistema imune inato e adaptativo, diferenciação celular, proliferação e sobrevivência em quase todos os organismos multicelulares.15,20 |
| BCL10 | O gene BCL10 (B‐cell CLL/lymphoma 10) codifica um membro de complexo proteico (CBM), que também contém adaptadores da família CARD, para recrutamento de caspases, como CARD9 e MALT1. O complexo CBM está envolvido na ativação do NFkB após a estimulação de vários receptores nas células linfoides, mieloides e epiteliais. BCL10 forma heterotrímeros com diferentes proteínas CARD em diferentes tipos de células.20 |
| MAPK | Gene MAPK (mitogen‐activatedproteinkinase) codifica enzima participante de vias de sinalização intracelular.20 |
| MALT1 | Gene (mucosa‐associated lymphoid tissue lymphoma translocation gene 1) ou paracaspase que codifica uma protease de cisteína do tipo caspase, essencial para a ativação do NFκB a jusante dos receptores da superfície celular.20 |
| CXCL | As quimiocinas são um grupo de moléculas pequenas (aproximadamente 8 a 14 kD) que regulam vários tipos de leucócitos por meio de interações com um subconjunto de receptores acoplados à proteína G 7‐transmembrana. São divididas em duas subfamílias principais, CXC e CC, com base no arranjo dos dois primeiros dos quatro resíduos de cisteína conservados; as duas cisteínas são separadas por um único aminoácido nas quimiocinas CXC e são adjacentes nas quimiocinas CC. As quimiocinas CXC são subdivididas em tipos ELR e não ELR, com base na presença ou ausência de uma sequência de glu‐leu‐arg adjacente e N‐terminal. Os tipos ELR são quimiotáticos para neutrófilos, enquanto os tipos não ELR são quimiotáticos para linfócitos.20 Por exemplo, a IL‐8 é conhecida como chemokine (C‐X‐C motif) ligand 8, CXCL8. |
| Inflamassoma | O sistema imune inato apresenta receptores de reconhecimento padrão (PRRs) para sequências moleculares específicas presentes em microorganismos e conservadas durante a evolução, denominadas padrão molecular associado a patógenos (PAMPs). Os receptores do tipo Toll (TLRs) e receptores de ligação a nucleotídio contendo domínio com sequência repetida de resíduos do aminoácido leucina (NLRs) são considerados os principais receptores de reconhecimento de PAMPs. TLRs também reconhecem moléculas endógenas denominadas padrão moleculares associado ao perigo (DAMPs) que são liberadas durante estresse e lesão celular. A ativação desses receptores aumenta a expressão de genes e fatores que controlam a produção de interferon tipo I e de citocinas proinflamatórias. Enquanto os TLRs são expressos na membrana celular, os NLRs são sensores citoplasmáticos que oligomerizam para formar um complexo proteico de ativação, que é o inflamassoma.46 |
Acredita‐se que um gatilho inicial seja a ativação de DC plasmocitoides, estimuladas por complexos de restos de DNA do hospedeiro e LL‐37, que são produzidos por queratinócitos após injúria. Catelicidinas humanas (hCAP18/LL‐37) são peptídios antimicrobianos; o LL‐37 ativo é liberado a partir da pró‐forma hCAP18 por meio de proteólise exercida pela proteinase interagem com receptores toll‐like para ativação de células dendríticas (fig. 1).23

As células epiteliais, incluindo os queratinócitos, são fontes de citocinas da família IL‐1 (IL‐1F), composta por 11 membros com sua nomenclatura mais comum (e nomes alternativos entre parênteses): IL‐1α, IL‐1β, antagonista do receptor da IL‐1 (IL‐1RN), IL‐18, IL‐33, IL‐36α (IL‐IF6), IL‐36β (IL‐1F8), IL‐36γ (IL‐1F9), IL‐36Ra (IL‐1F5), IL‐37 (IL‐1F7) e IL‐38 (IL‐1F10).24
Os receptores correspondentes são assim denominados: para IL‐36α e β, receptores IL‐1Rrp2 e IL‐1RAcp, expressos em monócitos, linfócitos T e B; para IL‐36γ, os receptores são os mesmos, mas expressos em queratinócitos e células epiteliais; para IL‐36Ra, os receptores são IL‐1Rrp2 e SIGIRR, expressos por queratinócitos, monócitos e células dendríticas.18 Os membros da IL‐36 usam o mesmo receptor IL‐36R; as três primeiras mostram níveis semelhantes de atividade agonista após a ligação, mas ligação com IL‐36Ra não inicia uma resposta de sinalização, portanto é considerado antagonista (fig. 2).23,24

A expressão de IL‐36γ foi localizada na camada granulosa da epiderme, especialmente em queratinócitos peripustulares. A expressão de IL‐36α também foi fortemente detectada nas camadas superficiais da epiderme na lesão da PPG e placas de psoríase.23
Assim como as famílias IL‐1, a IL‐36 requer clivagem peptídica N‐terminal para desencadear a atividade pró‐inflamatória, por ação de proteases derivadas de grânulos de neutrófilos, catepsina, elastase e proteinase‐3, aumentando sua atividade biológica em aproximadamente 500 vezes.18,25
Os neutrófilos infiltrados coram positivamente para catepsina G, elastase de neutrófilos e proteinase 3 na lesão de psoríase em placas e da psoríase pustulosa. Em estudos experimentais, a IL‐36α foi processada e ativada pela elastase de neutrófilos, mas não pela catepsina G, e seu processamento foi impedido pela serpina A1, um inibidor específico da elastase de neutrófilos. Já a IL‐36γ foi ativada pela catepsina G, mas não pela elastase, e foi inibida pela serpina A3, um inibidor específico da catepsina G. A catepsina S derivada de queratinócitos ou fibroblastos é uma enzima que cliva IL‐36γ e a transforma em sua forma ativa, IL‐36γ‐Ser18. É possível que a IL‐36γ‐Ser18 induza hiperqueratose e produção de CXCL8 e regule a produção por queratinócitos de CXCL1, CXCL10 e CCL20 (fig. 2).26
Outras enzimas participam do processamento endógeno dessas citocinas, como as caspases (proteases cisteína‐aspárticas), enzimas proteolíticas amplamente conhecidas por seu papel no controle da morte celular e inflamação. A caspase 1 está essencialmente envolvida na expressão gênica, e a caspase 3, na liberação de IL‐36γ. A caspase 14 é expressa na epiderme e tem papel primordial na corneificação e proteção das camadas subjacentes da pele.23,27
O estímulo in vitro de queratinócitos humanos com essas citocinas aumenta a expressão gênica de várias quimiocinas para macrófagos (CCL3, CCL4, CCL5, CCL2, CCL17 e CCL22), linfócitos T (CCL20, CCL5, CCL2, CCL17 e CCL22) e neutrófilos (CXCL8, CCL20 e CXCL1). Além dos queratinócitos, os monócitos e as células dendríticas mieloides (essas 10 vezes mais que os monócitos) expressam receptor para IL‐36.28 A presença de membros da IL‐36 aumenta significativamente a liberação de IL‐1β e IL‐6 pelas células dendríticas, macrófagos M2 humanos e células de Langerhans.29 Ocorre estímulo à maturação funcional dessas células e manutenção de um sistema autócrino.23
Níveis elevados de antagonista do receptor da IL‐36 neutralizam a reação pró‐inflamatória da IL‐36 na psoríase em placas e na psoríase pustulosa. A mutação da perda de função do gene IL‐36RN leva à liberação da sinalização da IL‐36, e os queratinócitos desses pacientes produzem níveis mais altos de CXCL8 em resposta à IL‐36.23
Demonstrou‐se também que a IL‐36 é capaz de ativar o endotélio vascular, levando a extravasamento plasmático significativo, o que resulta em edema acentuado da derme papilar, extravasamento de hemácias e de outras células, como eosinófilos.23,24
A expressão de citocinas relacionadas com Th17/Th1, como IL‐17A, IL‐22, IL‐23p19, IFN‐γ e IL‐18, é aumentada na psoríase em placas em comparação com PPG e com pele normal. A IL‐17A induz a expressão de IL‐36 mais intensamente nos queratinócitos humanos derivados da psoríase do que nos queratinócitos saudáveis.23
Manifestações clínicasO início do quadro tende a ser abrupto e explosivo, inicialmente com a presença de eritema e edema de extensão e grau variáveis, muitas vezes acometendo a região das grandes dobras (fig. 3). A pele está sensível e dolorosa. Dentro de horas surgem dezenas a centenas de pústulas estéreis não foliculares que se disseminam e podem confluir, formando lagos de pus (fig. 4). A erupção tende a ser generalizada, porém há predileção pelo tronco e pelos membros proximais (figs. 5‐7); as lesões podem surgir sobre pele aparentemente sã ou sobre lesões prévias de psoríase em placas (fig. 8). Enantema pustuloso da mucosa oral nos períodos de agudização e quadros persistentes de língua geográfica podem ocorrer, e os lábios podem apresentar descamação e exulceração. Pústulas subungueais podem estar presentes.30






Com a evolução do quadro, há dessecamento das pústulas, seguido de descamação escarlatiniforme, deixando a superfície lisa e brilhante (fig. 9). Comumente, persiste por poucas semanas, revertendo ao quadro anterior ou transformando‐se em psoríase eritrodérmica. As lesões tendem a regredir sem deixar sequelas. Raramente, cicatrizes hipertróficas ou queloides podem ocorrer. Episódios subsequentes de pustulização podem ocorrer, com intervalo de tempo variando de semanas a anos.6,30

As manifestações sistêmicas são frequentes e, muitas vezes, graves. A erupção pode ser acompanhada de sintomas sistêmicos como fadiga, mal‐estar, anorexia, náuseas, tremores e febre.8,31 Alterações oculares manifestas como conjuntivite, irite e uveíte também podem ser observadas. Artrite e osteomielite também foram relatadas.30
O quadro pode evoluir com complicações potencialmente graves, como superinfecção bacteriana, distúrbios metabólicos, hemodinâmicos e termorregulatórios, insuficiência renal, hepática e cardíaca, insuficiência respiratória aguda, colangite neutrofílica, pancreatite, choque asséptico e hipovolêmico e óbito. Tais consequências decorrem da quebra da barreira cutânea, vasodilatação e hipoalbuminemia. Febre e queda do estado geral estão habitualmente associadas. A ocorrência de colestase secundária à colangite neutrofílica bem como a síndrome da angústia respiratória aguda, se não diagnosticadas precocemente, também podem ter êxito fatal.4,32
Nos períodos entre os surtos de PPG, o indivíduo tende a permanecer livre das manifestações sistêmicas da fase aguda. Esses períodos de acalmia podem se estender por semanas ou anos. Quadros de psoríase em placas associados tendem a evoluir de modo independente das crises pustulosas.30,31
Em crianças com PPG, o curso tende a ser mais benigno, porém quadros graves e de êxito fatal são relatados.33 O manejo imediato e intensivo é mandatório, a fim de evitar complicações graves como superinfecções bacterianas, sepse, distúrbios metabólicos, hemodinâmicos e termoregulatórios.33
A psoríase pustulosa da gestação é manifestação bastante específica da PPG, antes denominada impetigo herpetiforme; a primeira descrição foi feita por Hebra, em 1872.5,34 Mais frequente no terceiro trimestre da gravidez, pode também ocorrer em fases precoces da gestação, no puerpério e até mesmo no período menstrual. Costuma se resolver com o final da gestação, mas pode recorrer em gestações subsequentes, com início mais precoce e quadro clínico mais grave a cada gestação. Associa‐se a maior risco de morbidade e mortalidade fetal secundário à insuficiência placentária.5,34 Clinicamente, consiste em erupção de pequenas pústulas sobre base eritematosa, em arranjo herpetiforme e disposição circinada na periferia das placas. No centro, as pústulas vão dessecando e deixando crostas. A erupção usualmente inicia nas áreas de flexuras, principalmente na região inguinal, com posterior disseminação.5,34 Febre, diarreia e náuseas podem estar presentes, bem como leucocitose com neutrofilia, aumento da velocidade de hemossedimentação (VHS), hipocalcemia e hipoalbuminemia, que pode levar a incontinência urinária e fecal, convulsões, tetanismo e óbito.35 O diagnóstico precoce e a instituição de monitorização materna e da vitalidade fetal são essenciais. Embora atualmente o prognóstico materno seja favorável mesmo nos casos complicados com delírio, convulsões e tetanismo secundários à hipocalcemia, o prognóstico fetal não é tão bom. Anormalidades fetais, prematuridade e morte neonatal secundários à insuficiência placentária podem ocorrer, mesmo nos casos bem controlados.5,34
Assim como no caso da PPG, a classificação da psoríase pustulosa da gestação é motivo de controvérsia. Embora seja usualmente considerada variante da psoríase pustulosa, alguns autores defendem sua classificação como entidade distinta relacionada à gestação, visto que a maioria das pacientes não apresenta história pessoal ou familiar de psoríase, a doença tem resolução com o término da gestação e recorre em gestações posteriores.5,34 Por outro lado, relatos do desenvolvimento de PPG em mulheres que tiveram a forma específica da gestação, anos após a remissão, favorecem a classificação da doença como forma de psoríase pustulosa.36
Critérios diagnósticosOs guias europeus de 2017 estabeleceram critérios diagnósticos para a PPG, incluindo a presença de pústulas primárias, estéreis, não foliculares e macroscópicas acometendo a pele não acral; pode ocorrer ou não na presença de psoríase em placas, de modo recorrente (mais de um episódio) ou persistente (duração de mais de três meses).37
Os guias japoneses de 2018 estabeleceram os seguintes parâmetros para o diagnóstico: a) sintomas sistêmicos como febre e fadiga; b) flushing extenso ou sistêmico acompanhado de pústulas estereis múltiplas que podem confluir para lagos de pus; c) na histopatologia, presença de pústulas neutrofílicas subcórneas caracterizadas como pústulas espongiformes de Kogoj; d) recorrências repetidas dos achados clínicos e histopatológicos anteriores. A presença de quatro dos critérios anteriores estabelece o diagnóstico definitivo de PPG, e a presença de dois ou três critérios, a suspeita diagnóstica.38
Fatores desencadeantesPacientes com psoríase em placas podem ter lesões pustulosas sobre as placas ou a distância em decorrência, sobretudo, de fatores agravantes como infecções principalmente do trato respiratório superior, queimaduras solares, irritantes locais como coaltar e antralina, estresse, gestação ou mesmo iniciar de forma idiopática. Uso de medicações como lítio, salicilato, alcatrão, cloroquina, betabloqueadores, indometacina dentre os anti‐inflamatórios não esteroides e, em especial, o uso e a descontinuação de corticoide sistêmico são clássicos agravantes de psoríase.39
A hipocalcemia pode ser causa ou consequência da psoríase pustulosa. Muitos estudos sugerem que a vitamina D desempenha um papel na diferenciação e proliferação celular da epiderme e a adesão celular precisa de caderinas, moléculas dependentes de cálcio. Há casos de hiperparatireoidismo e hipocalcemia com psoríase pustulosa que se reverteram apenas com a correção do distúrbio do cálcio.32,33
Dados do Registro Europeu de Reações a Fármacos (EuroSCAR) sobre risco de reações adversas cutâneas graves por fármacos com 97 casos de pustulose generalizada e 1.009 controles mostraram que os fármacos associados à PPG foram pristinamicina, ampicilina ou amoxicilina, quinolonas, (hidroxi) cloroquina, sulfonamidas, terbinafina e diltiazem. O quadro é dificilmente diferenciado da pustulose exantemática e generalizada aguda (PEGA).40 Há relatos sobre casos desencadeados por terbinafina, na forma de pustuloses generalizadas de novo, classificadas como PEGA, transformação pustulosa de psoríase em placas, PPG e pustulose palmoplantar.37,41
O uso do interferon para tratamento de diversas condições, como linfomas e hepatites, foi associado ao desencadeamento e à exacerbação de várias formas de psoríase, bem como o uso de imiquimode tópico, que pode aumentar a expressão de interferon na pele tratada e mesmo a distância.42
Em estudo retrospectivo com 102 pacientes com PPG aguda, os glicocorticoides sistêmicos foram imputados como gatilho em 44% dos casos, e a retirada abrupta de corticosteroides ou o uso de corticoide de depósito como única estratégia para tratamento da psoríase ou para tratar outra condição associada parecem ser a situação mais frequente.43
Choon et al. mostraram que infecções agudas são um fator desencadeante ou exacerbador em 16% dos pacientes, e que 38,5% apresentavam anticorpos antiestreptolisina. Wang et al., em estudo com 26 pacientes chineses, demonstraram que infecções foram responsáveis por desencadear a doença em 73% dos casos.43
Há casos de PPG sem manifestações prévias de psoríase em placas e em pacientes que não foram expostos aos fatores agravantes. Esses casos devem ser os mais relacionados às mutações genéticas descritas.9 Fototerapia, ustequinumabe, anti‐TNFα e até metotrexato, usados para tratamento de psoríase em placas, podem ser fatores de agravo e transformação para psoríase pustulosa.6,44
Diagnóstico diferencialO diagnóstico diferencial clínico se faz com outras dermatoses pustulosas, como a pustulose subcórnea de Sneddon Wilkinson, o pênfigo por IgA, a pustulose amicrobiana das dobras e, principalmente, a PEGA. A diferenciação entre PPG e PEGA pode ser difícil tanto clinicamente quanto na histologia, sobretudo quando há história de exposição a fármacos precedendo a erupção. Sugere‐se que a ausência de história pessoal ou familiar de psoríase, a resolução rápida com a suspensão dos agentes suspeitos, bem como a recorrência com a reexposição ao fármaco suspeito, associadas à presença de infiltrado eosinofílico, edema importante da derme superficial, vasculite, exocitose de eosinófilos, necrose de queratinócitos e ausência de alterações psoriasiformes na histologia, favoreçam o diagnóstico de PEGA.45
Por outro lado, uma série de doenças autoinflamatórias pode cursar com pústulas estéreis. Originam‐se de alterações na imunidade inata e da atividade excessiva e não controlada de inflamassomas46 – complexos proteicos que controlam a produção de citocinas pró‐inflamatórias. A função normal dos inflamassomas possibilita a produção ordenada de IL‐18 e, principalmente, de IL‐1, necessárias para a eliminação de patógenos e células cancerígenas. A participação dos inflamassomas foi descrita na síndrome de Sweet, vitiligo, hidradenite supurativa, dermatite atópica e psoríase. São classificadas como doenças autoinflamatórias monogênicas, um grupo de síndromes genéticas raras associadas à atividade da resposta imune inata com sinais recorrentes de inflamação sistêmica como febre, que surgem na infância e podem ter manifestações cutâneas. São exemplos a febre familiar do mediterrâneo e a síndrome de Muckle‐Wells. Doenças autoinflamatórias podem ter várias manifestações cutâneas, com pústulas disseminadas, à semelhança da psoríase pustulosa, urticas, paniculites, entre outras.47
Estudo sobre as várias possíveis mutações genéticas foi realizado em nove crianças de seis famílias que apresentaram osteomielite estéril neonatal, periostite e pustulose generalizada, e foi detectada uma mutação genética envolvendo o IL‐1RN, gene codificador do antagonista do receptor de IL‐1, que inibe IL‐1α e IL‐1β, altamente pró‐inflamatórias. Essa entidade foi denominada DIRA (deficiency of the IL‐1R antagonist), descrita em pacientes recém‐nascidos com quadro de osteomielite multifocal estéril, periostite, pústulas discretas a quadros ictiosiformes com úlceras orais e pittings ungueais. Isso despertou o interesse para estudos de outras condições dermatológicas por autoinflamação, como na síndrome SAPHO (sinovite, acne, pustulose e osteíte) e na PPG.17
Por outro lado, a mutação no gene IL‐36RN foi originalmente relatada em estudo de nove famílias da Tunísia com PPG. DITRA (deficiency of the IL‐36R antagonist) é o acrônimo de deficiência do antagonista do receptor da IL‐36, e é o achado em casos de PPG familiares ou esporádicos na Europa e Ásia. A DITRA foi inicialmente descrita em casos de PPG, e, posteriormente, com frequência menor, em PPP, ACH e PEGA, bem como relatos de casos e psoríase em placas com transformação pustulosa. Quadros associados a DITRA podem ocorrer de recém‐nascidos a adultos, e o quadro clínico pode ser grave com pustulose generalizada e sintomas sistêmicos, evoluindo para complicações da eritrodermia, sepse e risco de morte. A resposta aos tratamentos convencionais é insuficiente, e os pacientes deixam de responder aos imunobiológicos anti‐TNF‐α em curto espaço de tempo. Relatos demonstram que os fármacos anti‐IL‐17 e anti‐IL‐1 parecem ser mais eficazes; há pesquisas sobre anti‐IL‐36 para o tratamento dessas formas pustulosas (ver abaixo).12
HistopatologiaDe modo semelhante aos achados da psoríase em placas, observa‐se na PPG quadro de hipogranulose intercalada com paraqueratose (correspondente à epiderme suprapapilar), cones epiteliais alargados e regulares e afilamento suprapapilar da epiderme, aumento de vascularização da papila dérmica, bem como infiltrado linfoide perivascular na derme papilar inferior.1
São característicos os aglomerados de neutrófilos no estrato córneo (microabscesso de Munro), a presença de pústulas subcórneas e as pústulas espongiformes de Kogoj (fig. 10).1
. Hematoxilina & eosina, 100× e 400× (inset).")
Na histologia, a PPG deve ser diferenciada de outras dermatoses que podem cursar com pústula intraepidérmica, dentre elas a síndrome de Behçet, bromoderma, iododerma, candidose, sífilis secundária, dermatofitose, pustulose acral da infância, farmacodermias, reação à picada de insetos, impetigo, miliária cristalina, pênfigo foliáceo, pioderma gangrenoso, escabiose, síndrome da pele escaldada estafilocócica, pustulose amicrobiana das dobras e melanose pustulosa transitória neonatal.30
Nos casos de DITRA, foram detectadas as seguintes peculiaridades: camada córnea fina e compacta, acidofílica com hipogranulose difusa entre as pústulas; espongiose ao longo do estrato espinhoso; neutrófilos que migram para a epiderme; queratinócitos basais de aspecto regenerativo; edema acentuado da derme papilar com extravasamento de hemácias; eosinófilos no infiltrado inflamatório linfo‐histiocitário na derme papilar inferior.22
A diferenciação histopatológica com PEGA é difícil; a presença de queratinócitos apoptóticos e eosinófilos favorece o diagnóstico de PEGA. A presença de vasos tortuosos na derme superior parece favorecer o diagnóstico de PPG.45
Importante lembrar que, na investigação de erupções pustulosas, a imunofluorescência direta deve ser realizada para afastar formas de pênfigo, como os causados por depósito de IgA intercelular, ou variantes de pênfigo foliáceo e vulgar por IgG anti‐desmogleína 1 e 3.48
Avaliação laboratorialAvaliação laboratorial é necessária para verificar a gravidade do quadro e diagnosticar possíveis complicações. As alterações laboratoriais estão ligadas ao grau de inflamação e comprometimento sistêmico.
Podem ocorrer distúrbios hidroeletrolíticos e hipovolemia, hipoalbuminemia, elevação das enzimas hepáticas, elevação de bilirrubinas e leucocitose com neutrofilia e linfopenia. Elevação das provas de atividade inflamatória são frequentes. Hipocalcemia pode ocorrer como reflexo da hipoalbuminemia, mas geralmente é assintomática, e a dosagem de cálcio iônico é normal.
Recomenda‐se realizar hemograma completo, dosagem de enzimas hepáticas, ureia, creatinina, proteína C reativa, VHS, dosagem de cálcio e albumina. Culturas de sangue e urina são necessárias para descartar infecções.5
Os testes genéticos discutidos a seguir não são indicados de rotina, pois ainda apresentam disponibilidade limitada e custo elevado.
Triagem genética na investigaçãoTrês mutações são responsáveis por menos de 30% dos casos de PPG: IL‐36RN, CARD14 e AP1S3. Apesar de os exames genéticos para mutações não serem rotineiramente indicados em razão da dificuldade de acesso e do alto custo, as mutações no IL‐36RN são cada vez mais usadas para auxiliar no diagnóstico de PPG.
Tais mutações são associadas à idade mais precoce de início em todos os subtipos de psoríase pustulosa, evolução mais grave, inflamação generalizada e diferentes respostas ao tratamento. Twelves et al. recomendam que todos os pacientes com início de PPG antes dos 30 anos devam ser rastreados quanto a mutações no IL‐36RN.49
TratamentoAlém de rara, a PPG apresenta evolução característica em surtos com possível remissão espontânea. Esses fatores dificultam a execução de ensaios clínicos randomizados e a elaboração de esquemas de tratamento padronizados e algoritmos de tratamento.9
A extensão e a gravidade da PPG demandam pronta intervenção, internação e, não raramente, suporte de unidade de terapia intensiva.9
AcitretinaOs retinoides são os agentes sistêmicos mais antigos estudados no tratamento da PPG. O etretinato foi o primeiro retinoide testado e teve sua eficácia demonstrada em séries de casos; no entanto, não é considerado opção terapêutica por não estar mais disponível.9
A acitretina teve sua eficácia demonstrada em estudos retrospectivos9 conduzidos na Europa4 e na Ásia. Augey et al. avaliaram 99 pacientes com PPG em 46 centros da França, nos quais a acitretina foi utilizada em 89% dos pacientes como tratamento de primeira linha, e foi considerado pelos autores o tratamento sistêmico mais efetivo.4 No segundo estudo, conduzido por Choon et al., foram avaliados 102 pacientes com PPG; desses, 52 foram respondedores ao retinoide sistêmico.
A dose recomendada varia entre 0,75 a 1mg/kg/dia, e os pacientes geralmente respondem em 7 a 10 dias.50 Por seu potencial teratogênico, não deve ser utilizada em gestantes ou mulheres em idade fértil.
MetotrexatoA eficácia do metotrexato na PPG foi evidenciada em dois estudos retrospectivos, o primeiro com 24 pacientes e o segundo com 41. O etotrexato foi considerado eficaz em 76,2% e 80% dos pacientes, respectivamente, porém com análises de desfechos distintos.50 As doses sugeridas são semelhantes às empregadas na psoríase em placas, variando entre 15 e 25 mg/semana.
CiclosporinaAs evidências que sustentam o uso da ciclosporina são, em grande parte, provenientes de relatos de caso.50 Em estudo retrospectivo, 66 pacientes foram tratados com ciclosporina em diferentes centros do Japão. A ciclosporina foi considerada eficaz em 71,2% dos pacientes.50
FototerapiaNão existem evidências para recomendar o uso da fototerapia no surto (fase aguda) da PPG. Existem relatos da eficácia da fototerapia PUVA para ser utilizada na manutenção, após controle do surto agudo.9
BiológicosInibidores do TNF‐alfaDesde a aprovação dos inibidores do TNF‐alfa para psoríase em placas, diversas publicações, em sua maioria relatos de casos e séries de casos, relataram a eficácia dessa classe terapêutica na PPG.50
Em revisão publicada em 2018 por Boehner et al., foram identificados na literatura 55 casos de PPG tratados com agentes anti‐TNF‐alfa, a maior parte (29 pacientes) com infliximabe. Do total de pacientes tratados, 58% apresentaram remissão total e 28%, resposta parcial. Importante lembrar que os anti‐TNF‐alfa, embora venham sendo empregados no tratamento da PPG, podem ser responsáveis pelo desencadeamento de surtos de PPG aguda, considerada uma reação paradoxal.9
InfliximabeA evidência para recomendar o uso de infliximabe no tratamento da PPG vem de relatos de caso de sucesso e série de casos.50 Estudo realizado em pacientes japoneses com várias formas de psoríase incluiu sete pacientes com PPG, e as taxas de resposta ficaram em torno de 70%.50 Seu emprego na PPG foi motivado pelo rápido início de ação. O Guia de Tratamento de Psoríase Pustulosa da National Psoriasis Foundation (2012) considerou o infliximabe o tratamento de primeira linha, posicionando etanercepte e adalimumabe como tratamentos de segunda linha.9
AdalimumabeEm estudo aberto, multicêntrico, de 52 semanas, Morita et al. avaliaram a eficácia e a segurança do adalimumabe em 10 pacientes japoneses com PPG; a taxa de remissão completa foi de 50% em duas semanas e de 70% em 16 semanas. Dos sete pacientes que apresentaram remissão completa, houve escalonamento de dose para 80 mg a cada duas semanas. Embora seja considerado tratamento de segunda linha no Guia de Tratamento de Psoríase Pustulosa da National Psoriasis Foundation (2012), é o primeiro anti‐TNF‐alfa avaliado em ensaio clínico, mostrando ser eficaz e seguro no manejo da PPG.9
EtanercepteÉ o anti‐TNF‐alfa com menor evidência no tratamento da PPG, embora também com publicações de relatos de caso de sucesso terapêutico.50 No Guia de Tratamento de Psoríase Pustulosa da National Psoriasis Foundation (2012), aparece como segunda linha de tratamento.9
Ustequinumabe (anti‐IL‐12/IL‐23)Embora sejam relatados casos de ustequinumabe desencadeando surtos de PPG,9 existem relatos de caso e série de casos com sucesso terapêutico, inclusive de sua utilização bem‐sucedida no manejo da PPG desencadeada pelos inibidores do TNF‐alfa.50 Arakawa et al. publicaram uma série de quatro casos de PPG refratários a vários tratamentos prévios (incluindo anti‐TNF‐alfa) que apresentaram remissão completa com o uso de ustequinumabe.9
Anti‐IL‐17Nos últimos anos, foram realizados estudos abertos avaliando a eficácia e segurança dos biológicos anti‐IL‐17 no tratamento da PPG. Em estudo aberto com 12 pacientes tratados com secuquinumabe, houve boa resposta em 83% dos pacientes.9 O brodalumabe foi avaliado em estudo aberto que incluiu 12 pacientes, no qual foi observada 83% de melhora ou remissão em 12 semanas.9,50 Resultados semelhantes foram obtidos com ixekizumabe em dois estudos que incluíram pacientes com PPG.9 Os resultados com anti‐IL‐17 são promissores, mas ainda são necessários estudos com amostras maiores, comparação com grupo controle e estudos em populações não asiáticas.9
Anti‐IL‐23Os resultados iniciais promissores com os anti‐IL‐17 motivaram a avaliação dos biológicos anti‐IL‐23, que também bloqueiam o eixo IL‐23/Th17, no tratamento da PPG. Em estudo aberto com guselcumabe, foram tratados 10 pacientes com PPG, e observadas taxas de sucesso de 50% em uma semana e 100% em 52 semanas para os oito pacientes que concluíram o estudo.9
Terapia alvo contra citocinas do eixo IL‐1/IL‐36As citocinas da família IL‐1, em particular a IL‐36, desempenham importante papel na imunopatogenia da PPG,9 o que tem motivado os pesquisadores a avaliar terapias que têm essas citocinas como alvo.
Relatos de caso têm demonstrado eficácia do anakinra (antagonista do receptor de IL‐1) e do canaquinumabe e gevoquizumabe (anticorpos monoclonais anti‐IL‐1β).9,32
Novos fármacos que têm como alvo o receptor de IL‐36, anti‐IL‐36R, estão em fase de desenvolvimento (ANB019 e BI655130/spesolimab), e os resultados de fase 1 são promissores. Já estão em andamento estudos de fase 2 e 3.9
Guias de tratamentoNa Europa e nos EUA não existe aprovação de nenhum agente terapêutico para PPG até o presente momento. No Japão, há aprovação para o uso dos agentes imunobiológicos anti‐IL‐17 (secuquinumabe, ixequizumabe e brodalumabe) e anti‐IL‐23 (guselcumabe).9
Em 2012, a National Psoriasis Foundation publicou um guia de tratamento da PPG recomendando acitretina, ciclosporina, metotrexato e infliximabe como opções de tratamento de primeira linha para a PPG em adultos.9
Nos casos de impetigo herpetiforme, a condição crítica do binômio materno‐fetal e a possibilidade de teratogenicidade causada pelos fármacos são desafios enfrentados pelo dermatologista. Embora sejam relatadas intervenções medicamentosas, o nível de evidência é baixo e não existem estudos controlados para o tratamento do impetigo herpetiforme. Não se dispõe, até o momento, de guias de tratamento específicos para impetigo herpetiforme.34
PrognósticoA PPG apresenta curso variável e imprevisível; a maior parte dos pacientes apresenta doença recidivante. O tempo entre as crises é variável, podendo ou não haver regressão completa das lesões entre as mesmas. A gravidade das crises também pode variar no mesmo paciente.
O aparecimento das pústulas pode se dar sem grandes sinais de inflamação sistêmica ou estar associado a quadros inflamatórios intensos, com rápida progressão e risco de morte, indicando a necessidade de internação em unidades de terapia intensiva. Dados sobre as taxas de mortalidade são limitados e foram reportados entre 3 e 7% dos casos de PPG.33
Identificar a presença de infecção costuma ser difícil em decorrência do intenso processo inflamatório; decidir sobre o equilíbrio entre o uso de imunossupressores e antibióticos é um desafio. Durante a avaliação na crise, deve‐se buscar o reconhecimento precoce dos pacientes de maior risco e com potencial necessidade de internação. A vigilância criteriosa dos sintomas e da evolução clínica é fundamental para que se possa intervir precocemente e tentar evitar complicações graves.
Por ser uma doença incurável, o desenvolvimento de medicações capazes de evitar as crises ou mesmo de aumentar o intervalo entre as mesmas poderá melhorar o prognóstico e a qualidade de vida dos pacientes com PPG.5,49
Conclusão e perspectivas futurasO manejo da enfermidade ainda é um desafio na prática clínica da dermatologia por se tratar de uma dermatose grave para a qual existem poucos guias de tratamento padronizados. A raridade da doença e sua evolução característica (surtos com períodos de remissão) limita a execução de protocolos de pesquisa de melhor qualidade para avaliação da terapêutica.
Embora as evidências científicas ainda sejam escassas, o cenário parece estar em transformação graças a avanços do conhecimento nas áreas da genética e imunologia ocorridos nos últimos anos e o advento da terapia imunobiológica. A melhor caracterização genotípica e imunopatogênica das diferentes variantes pustulosas da psoríase tem possibilitado o desenvolvimento de novos alvos terapêuticos.
Suporte financeiroNenhum.
Contribuição dos autoresMarcelo Arnone: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Renata Ferreira Magalhães: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Ricardo Romiti: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
André Luis da Silva Hirayama: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflito de interessesRicardo Romiti tem conflito de interesses com os seguintes laboratórios, atuando como consultor e/ou palestrante: Teva, Abbvie, Boeringer‐Ingelheim, Janssen, Eli‐Lilly, Leo Pharma, Novartis, PPfizer, UCB Biopharma.
André L. S. Hirayama tem conflitos de interesse com os seguintes laboratórios, trabalhando como palestrante, pesquisador e em outros como consultor: Abbvie, Boeringer‐Ingelheim, Novartis, Janssen, Eli‐Lilly.Marcelo Arnone tem conflitos de interesse com os seguintes laboratórios, trabalhando como palestrante, pesquisador e outros como consultor: Abbvie, Boeringer‐Ingelheim, Novartis, Janssen, Eli‐Lilly, Leo Pharma e UCB Biopharma.
Renata F. Magalhães tem conflitos de interesse com as seguintes instituições, atuando como palestrante e em outras como pesquisadora: Abbvie, Novartis, Janssen‐Cilag, Eli‐Lilly e Unicamp.
Como citar este artigo: Romiti R, Hirayama ALS, Arnone M, Magalhães RF. Generalized pustular psoriasis (von Zumbusch). An Bras Dermatol. 2022;97:63–74.
Trabalho realizado no Departamento de Dermatologia, Hospital das Clínicas, Universidade de São Paulo, SP, Brasil e na Disciplina de Dermatologia, Faculdade de Ciências Médicas da Unicamp, Campinas, SP, Brasil.
