










Cutaneous mucinoses are a heterogeneous group of dermatoses in which excess deposition of mucin in the dermis gives the skin a waxy appearance, with papules and plaques that can vary from self-healing mucinosis to even disrupting the normal shape of a patient’s face, conferring a leonine facies, or be part of life threatening diseases like scleromyxedema. This review will describe the most recent classification on lichen myxedematosus in the generalized (scleromyxedema) and the localized forms, as well as the different organ systems involved in scleromyxedema, diagnostic workup, current management, and prognosis.
Cutaneous mucinoses are a heterogeneous group of skin disorders characterized by an abnormal dermal deposition of mucin. Mucin is composed of glycosaminoglycans, hyaluronic acid, and dermatan sulfate. It is a component of the extracellular matrix and is produced by fibroblasts. Mucin can absorb 1,000 times its own weight in water, and its main function is to maintain the salt and water balance within the dermis.1
The cause of its abnormal cutaneous deposition remains unclear, although there are diverse proposed physiopathogenic mechanisms, including paraproteinemia and serum factors that could promote mucin production within the skin.
This article describes lichen myxedematosus (LM), a cutaneous mucinosis first reported as a systemic disorder associated with paraproteinemia and extracutaneous involvement. A recent classification by Rongioletti et al. includes a generalized papular and sclerodermoid presentation (scleromyxedema), as well as localized, atypical, and intermediate forms with or without systemic involvement.2
HistoryScleromyxedema was a term first introduced by Gottron in 1954,3 giving credit to his former teacher Arndt for the original description of a chronic dermatosis in a female patient. Nevertheless, in 1908 Dubreuilh had previously described an identical dermatosis under the name of fibromes miliaries folliculaires: sclerodermie. Later in 1953, Montgomery and Underwood distinguished scleromyxedema from scleroderma and generalized myxedema, describing four different clinical patterns, a generalized lichenoid eruption, a discrete papular form, a localized or generalized lichenoid plaque, and an urticarial plaque type.4
DefinitionLM represents a group of rare cutaneous diseases characterized by papules, nodules, and plaques of mucin deposition that give the skin a waxy appearance, with a chronic course and a poor therapeutic response.5
PathogenesisThe pathogenesis of scleromyxedema is unknown. The most accepted hypothesis is that circulating cytokines such as IL-1, TNF-alpha, and TGF-beta, which stimulate glycosaminoglycan synthesis and proliferation of fibroblasts, could play a role.6In vitro serum of patients with scleromyxedema can stimulate synthesis of DNA by fibroblasts. Although monoclonal gammopathy occurs, more often IgG is observed in patients with disseminated disease, and levels of the paraproteinemia do not correlate with disease severity. Moreover, tissue mucin deposition in autopsies does not correlate with the clinical findings.7
Diagnostic criteria and classificationThe latest classification for LM by Rongioletti et al. distinguishes between three different subgroups of LM, each with different diagnostic criteria: generalized LM (scleromyxedema), a localized form, and an atypical variant which shares characteristics of the first two, but fulfilling diagnostic criteria for neither of them.5
Diagnostic criteria for generalized LM or scleromyxedema are as follows: (a) microscopic triad of mucin deposition, fibroblast proliferation, and fibrosis; (b) monoclonal gammopathy, predominantly IgG λ and less frequently IgG κ; (c) absence of thyroid disease.
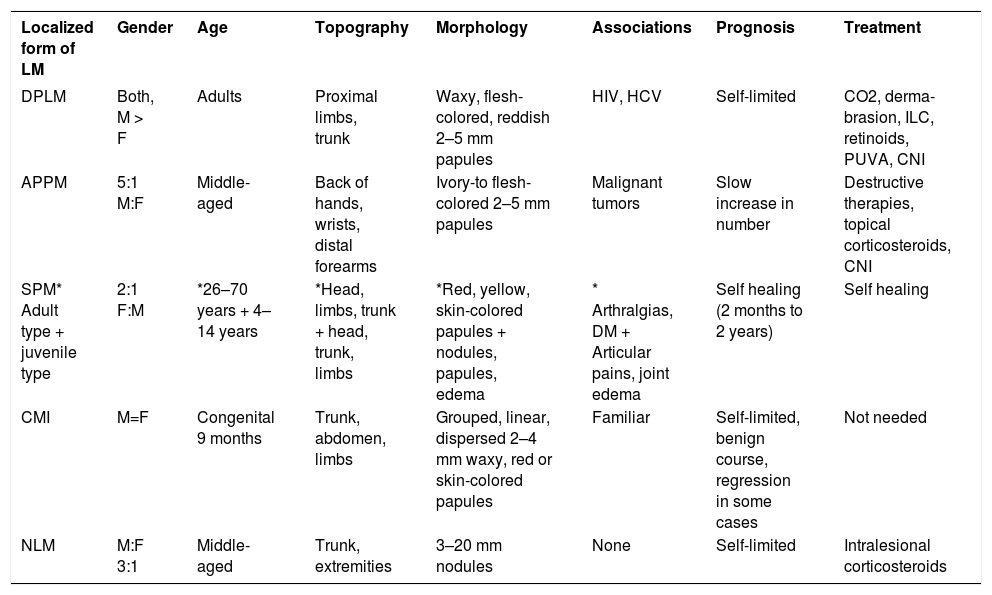
For the localized LM variant, five subtypes can be distinguished: (a) discrete papular LM; (b) acral persistent papular mucinosis; (c) self-healing papular mucinosis; (d) papular mucinosis of infancy; and (e) nodular LM. All of them must have the clinical and histopathological characteristics of LM but without paraproteinemia, systemic involvement, or thyroid disease (Figure 1).

Atypical subtypes do not fulfill the mentioned criteria and share characteristics of both scleromyxedema and localized LM; the latter is also known as papular mucinosis.5
Recently, Nofal et al. proposed a grading system for LM based on the extension of cutaneous involvement and the presence of systemic manifestations: G1 (mild), limited pure cutaneous involvement; G2 (moderate), extensive pure cutaneous involvement; or G3 (severe), limited or extensive cutaneous involvement plus systemic manifestations.8
ScleromyxedemaScleromyxedema is a rare skin disorder clinically characterized by a disseminated eruption of 2–3 mm waxy, firm, dome-shaped or flat-topped papules and nodules that may coalesce to form plaques involving the head, neck, trunk, and extremities. The scalp and mucosa are generally not affected.9 Papules are commonly arranged in a linear array (Figure 2) and the surrounding skin has a sclerodermoid appearance. There is no significant predominance by gender, and it is most common in adults between the fifth and sixth decades of life.10

Mucin deposition within the dermis is responsible for the cutaneous findings. A sclerodermoid eruption with multiple papules, edema, and erythema - as well as papular induration over the ears and glabella - confer a leonine facies (Figure 3).11,12 Other skin findings include the doughnut sign, which is a central depression surrounded by an elevated rim on extensor proximal phalangeal joints. Deep furrowing can also be evident on the back. Skin thickening in chronic scleromyxedema can result in diminished articular movement and difficulty when opening the mouth. Pruritus and dysesthesia are also common complaints.10,11

By definition, a monoclonal gammopathy, more commonly IgG λ and systemic involvement such as neurological, rheumatoid, cardiac, pulmonary, gastrointestinal, hematologic, and ocular manifestations must be present in scleromyxedema.
Extracutaneous involvement has been reported in 70% to 77% of patients.
Neurological involvement in scleromyxedemaParaproteinemia with or without hyperviscosity might explain neurologic manifestations such as encephalopathy, seizures, cerebrovascular events, transient focal neurologic disturbances, and peripheral neuropathy observed in scleromyxedema.13,14 Mucin deposition in the brain has not been demonstrated to be responsible for these. Peripheral neuropathy in scleromyxedema remains unclear, and carpal tunnel syndrome has been explained as a deposition of mucin within the wrist or secondary to direct median nerve toxicity.3
Other neurological conditions related to scleromyxedema are considered iatrogenic, secondary to treatments with cytotoxic agents.15,16
Dermato-neuro syndrome, presenting with fever, seizures, and coma, along with a flu-like prodrome, is a rare complication of scleromyxedema. The pathophysiology is poorly understood, but monoclonal gamma paraproteinemia and blood hyperviscosity have been postulated as causes for this rare complication.17,18
Treatment is empirical, although IVIG has been used successfully in combination with plasmapheresis. Other treatments described in the literature include thalidomide, corticosteroids, and melphalan.15,19
Cardiac involvement in scleromyxedemaCardiac manifestations in scleromyxedema include myocardial ischemia, and congestive and inflammatory cardiomyopathies.3,11,12
Pulmonary involvementThe most common pulmonary finding in scleromyxedema is dyspnea. Obstructive and restrictive pathologies are the main causes. Atherosclerosis of the larger branches of the pulmonary arteries and congestion have also been found.11,20,21
Gastrointestinal involvementThe most common gastrointestinal finding in patients with scleromyxedema is hypomotility of the esophagus and dysphagia.11
Musculoskeletal involvementProximal muscle weakness is the most common musculoskeletal finding in scleromyxedema, as well as arthralgias, myositis, and fibromyalgia.
Hematologic involvementParaproteinemia is commonly encountered, mainly IgG λ. Multiple myeloma has been associated with monoclonal gammopathy and patients with scleromyxedema have been reported to progress to multiple myeloma in up to 10% of cases. Hodgkin and non-Hodgkin lymphomas, as well as myelodysplastic syndromes with progression to myeloid leukemia have also been reported.21–23
Other systemic manifestations reported in the literature include macular edema, Raynaud’s phenomenon, and Sjogren’s syndrome.11
DiagnosisBesides a thorough patient history and characteristic physical and histologic findings, serum protein immunoelectrophoresis and immunofixation to evaluate for monoclonal gammopathy and thyroid studies to rule out myxedema of thyroid disease are required for adequate diagnosis. Additional studies to evaluate systemic disease such as an echocardiogram or hepatic ultrasound may be necessary.
Localized forms of LMDiscrete papular LMDiscrete papular lichen myxedematosus (DPLM) is a rare entity more commonly found in male patients and is strongly associated with HIV. An overstimulation of fibroblasts is present, leading to dermal mucinosis.24 Hepatitis C infections have also been found in Japanese patients with DPLM.
Cutaneous findings include 2–5mm bilateral, symmetrical, red-to-violaceous papules (Figure 4) affecting the trunk (Figures 5 and 6) and limbs, sparing the face.25 Progression occurs slowly, without systemic involvement, and evolution to scleromyxedema has never been documented. The prognosis is good, although spontaneous remission is rare.26,27
Acral persistent papular mucinosis


According to its name, acral persistent papular mucinosis (APPM) is a form of lichen myxedematosus (LM) in which waxy, skin-colored papules are found on the extensor surfaces of the hands, wrists, and forearms. The etiology is unknown; systemic involvement is absent and spontaneous remission does not occur.5,28
Self-healing papular mucinosisSelf-healing papular mucinosis (SPM) is a rare variant of localized LM with eruption of waxy papules, with no systemic involvement. There are few reports in the literature of this variant. It has similar clinical characteristics as DPLM but remission occurs spontaneously. Resolution has been reported from two months to 14 months, with no sequelae.5,29
Cutaneous mucinosis of infancyCutaneous mucinosis of infancy (CMI) is another variant of localized LM. It may be considered a pediatric variant of DPLM or APPM.30 It consists of erythematous-to-skin-colored papules < 1cm in diameter, more commonly found on the trunk and upper extremities.31 Different patterns have been described, such as symmetrical, densely grouped, linear, and localized, as well as a more generalized form with no systemic involvement (Figures 7 and 8).


This condition may be congenital or acquired early in infancy, and it must be distinguished from a mucinous naevus. Spontaneous regression is uncommon, although there are reports of this atypical course of CMI.32
Nodular LMNodular lichen myxedematosus (NLM), is characterized by hard, skin-colored nodules usually > than 6 mm with or without papules, located on the limbs and trunk. Very few cases of this type of LM have been reported, and the age of onset seems to be earlier compared to APPM and DPLM. There were no symptoms associated in four reported cases (Table 1).33–41
Localized forms of LM
| Localized form of LM | Gender | Age | Topography | Morphology | Associations | Prognosis | Treatment |
|---|---|---|---|---|---|---|---|
| DPLM | Both, M > F | Adults | Proximal limbs, trunk | Waxy, flesh-colored, reddish 2–5 mm papules | HIV, HCV | Self-limited | CO2, derma-brasion, ILC, retinoids, PUVA, CNI |
| APPM | 5:1 M:F | Middle-aged | Back of hands, wrists, distal forearms | Ivory-to flesh-colored 2–5 mm papules | Malignant tumors | Slow increase in number | Destructive therapies, topical corticosteroids, CNI |
| SPM* Adult type + juvenile type | 2:1 F:M | *26–70 years + 4–14 years | *Head, limbs, trunk + head, trunk, limbs | *Red, yellow, skin-colored papules + nodules, papules, edema | * Arthralgias, DM + Articular pains, joint edema | Self healing (2 months to 2 years) | Self healing |
| CMI | M=F | Congenital 9 months | Trunk, abdomen, limbs | Grouped, linear, dispersed 2–4 mm waxy, red or skin-colored papules | Familiar | Self-limited, benign course, regression in some cases | Not needed |
| NLM | M:F 3:1 | Middle-aged | Trunk, extremities | 3–20 mm nodules | None | Self-limited | Intralesional corticosteroids |
Source: Swapna, et al. 2016,36 Laura Atzori 2014,37 Ko-todziejczyk, Beata 2017,38 Luo, D 2011,39 Alvarez-Garrido 2014,40 Gomez Sanchez, Maria Encarnacion 2016.41 DPLM, discrete papular lichen myxedematosus; APPM, acral persistent papular mucinosis; SPM, self-healing papular mucinosis; CMI, cutaneous mucinosis of infancy; NLM, nodular lichen myxedematosus; ILC, intralesional corticosteroids; CNI, calcineurin inhibitor; DM, diabetes mellitus
Mucin is a normal component of the dermal connective tissue produced by fibroblasts and mast cells. In LM, this hyaluronic acid complex is increased, and because mucin is hygroscopic, which means it holds water, the connective tissue is swollen and excessive collagen is produced.42
When suspecting cutaneous mucinosis by clinical or histologic findings, special stains such as Alcian blue, toluidine blue, or colloidal iron should be requested (Figure 9).1

Alcian blue is a water-soluble polyvalent dye which stains both sulfated and carboxylated mucopolysaccharides, as well as sialomucins (glycoproteins), resulting in a blue stain in histopathology. Toluidine blue is an acidophilic metachromatic dye that stains acidic tissue components attached to glycosaminoglycans in mast cells or connective tissue, displaying a purple-to-red color. Colloidal iron stains mucin because low pH colloidal ferric irons are absorbed by carboxylated and sulfated mucosubstances; therefore, the Prussian blue reaction is used to demonstrate iron bound to tissue, revealing a deep blue color. Mucin is hyaluronidase sensitive and PAS negative; additionally, monoclonal antibodies to hyaluronan may also be used to detect mucin deposition in the dermis.
In LM, as previously mentioned, there is a triad of histologic features that comprises diffuse mucin deposition, proliferation of irregularly arranged fibroblasts, and increased collagen deposition. The epidermis may be normal or thinned in chronic lesions, secondary to the pressure of the underlying mucin. Follicles may be atrophic and a mild superficial perivascular lymphoplasmacytic infiltrate is often present.
The pattern and location of mucin deposition within the dermis are the main clues to histopathological diagnosis, as well as other additional findings. A diffuse pattern of mucin deposition is mostly seen in scleromyxedema, while a focal pattern is observed in localized LM. Mucin deposition in localized, and generalized forms of LM may be found in upper or mid dermis.1,5
Reflectance confocal microscopy (RCM) is an advanced imaging technique that provides dermatologists with a non-invasive tool to observe the interior structures of the skin. In a recent study, RCM was used to diagnose two patients with focal dermal mucinosis, and a correlation between RCM and histopathology was made. In both patients, mucin deposits within the dermal stroma were consistent with dark spaces between thin collagen fibers. In addition, spindle-shaped fibroblasts within the mucinous areas – with fusiform-to-dendritic structures – were observed to be thicker than the surrounding fibers. Proliferation of dermal capillaries was recognized as increased blood flow during real-time or video-mode imaging. Although the number of patients in this study was small, and more studies are needed to standardize patterns on RCM in dermal mucinoses, RCM may be a promising method for diagnosis.43
TreatmentBecause of the rarity of these diseases, there is no standardized consensus for their therapeutic approach. Several modalities have been used, with variable responses.
Some types of localized LM resolve spontaneously, such as self-healing papular mucinosis, rare cases of CMI, and DPLM. APPM does not exhibit regression, and despite the benign course of the disease, patients frequently seek cosmetic treatment. More severe presentations such as scleromyxedema must be promptly treated to prevent progression to a severe or even fatal outcome.
Melphalan was previously considered as first-line treatment for scleromyxedema in order to target the plasma cell dyscrasia. Favorable clinical responses were observed in some patients; nevertheless, it was responsible for 30% of the deaths recorded in scleromyxedema associated to hematologic malignancies and in-fections.44 Other chemotherapeutic agents alone or combined have been used, with variable outcomes. Cyclophosphamide and methotrexate have been used, with poor response.45
Thalidomide has been used alone or in combination with other systemic agents, with variable results.44,46,47 Improvement in symptoms such as pruritus and skin induration can be achieved with the use of thalidomide.48 One series of three patients previously unsuccessfully treated showed a marked response when thalidomide was started.36
Hydroxychloroquine along with PUVA and thalidomide were ineffective in another report.49 The combination of vincristine, idarubicin, dexamethasone, and thalidomide was reported to be successful in one patient with scleromyxedema, although thalidomide had to be interrupted due to neuropathy.22
Oral retinoids have also been used with partial response.50,51
Cyclosporine was effective in a patient previously unresponsive to photochemotherapy and prednisolone followed by plasmapheresis. The authors suggested a reduction in IL-6, induction of fibroblast proliferation, and production of protein M as cyclosporine’s mechanism of action.45
Currently, the most promising treatment for scleromyxedema is IVIG, although the exact mechanism is not fully understood. Immunomodulatory processes have been postulated, including functional blockade of Fc receptors on splenic macrophages, inhibition of complement-mediated damage, modulation of the production of cytokines and cytokine antagonists, neutralization of circulating autoantibodies by anti-idiotype antibodies in IVIG, neutralization of pathogens involved in the induction of autoimmune disease, CD95 blockade, and inhibition of apoptosis.20,52,53,54 Different protocols have been reported for the use of IVIG in scleromyxedema. A full-dose regimen of 2g/kg and a low-dose regimen of 0.4–0.5g/kg have been used. Nearly all reports using IVIG have demonstrated at least a partial response in skin manifestations and internal organ involvement.55 Serum monoclonal gammopathy does not seem to regress with IVIG treatment, even though clinical manifestations of the disease improve dramatically.
Bortezomib, a reversible proteasome inhibitor used as an antineoplastic drug in diseases like multiple myeloma and mantle cell lymphoma among other types of malignancies,56 has shown a good response in refractory scleromyxedema when used in combination with other therapies such as corticosteroids, and remission was achieved even after bortezomib was stopped.46,57
Autologous and allogenic stem cell transplants are both options in patients where other treatments have failed or in severe and refractory disease, and although relapses have been reported, most of the patients reported in the literature were previously treated with the multiple treatments described above.19,58,59
Patients with the localized variants of LM must be counseled about the benign course of their disease. Different therapeutic approaches can be used in the localized variants, including topical therapies, destructive modalities, and intralesional steroids.10 Among topical therapies, tacrolimus 0.1% is suggested to have the best response by inhibiting TNF-α and TGF-ß, thus reducing the synthesis of glycosaminoglycans by fibroblasts.
Complete resolution of LM nodules has been reported with intralesional triamcinolone acetonide 0.02–0.05 mL (8mg/mL).35
Destructive modalities including CO2 laser, Erbium-YAG laser, cryotherapy, electrofulguration, and electrocoagulation are suitable for the localized forms for cosmetic reasons.28
In HIV patients with associated DPLM, full resolution of papules and plaques has been reported after antiretroviral therapy.60
Final ConsiderationsLM represents a rare group of cutaneous mucinoses with excessive deposition of mucin in the dermis. Although the exact mechanism by which this occurs has yet to be elucidated, the most accepted hypothesis is that circulating cytokines and monoclonal gammopathy stimulates fibroblast growth.
The most accepted classification differentiates localized forms from disseminated LM or scleromyxedema, where systemic involvement featuring neurologic, cardiac, hematologic, gastrointestinal, and muscular disease can be life-threatening.
The diagnosis must be confirmed with histopathology, special stains for mucin deposition must be requested, and the presence of systemic disease should be assessed.
As of today, no clinical guidelines have been published for the treatment of localized or disseminated forms.
AcknowledgmentAnatomic Pathology Service, Hospital Universitario, Universidad Autónoma de Nuevo León for the microscopic images.
Financial support: None.
Conflict of Interest: None.
