








Epidermólise bolhosa (EB) é um grupo de doenças hereditárias raras, caracterizadas por fragilidade da pele e mucosas. Dados epidemiológicos de EB no Brasil são escassos.
ObjetivosDescrever aspectos epidemiológicos de pacientes com EB diagnosticados no Departamento de Dermatologia de um hospital terciário, de 2000 até 2022.
MétodosEstudo observacional e retrospectivo foi conduzido por meio da análise de prontuários. Dados avaliados incluíram forma clínica, sexo, história familiar, consanguinidade, idade ao diagnóstico, idade atual, tempo de seguimento, comorbidades, exame histopatológico e imunomapeamento, presença de nevos EB e de carcinomas espinocelulares (CEC), causa e idade da morte.
ResultadosDos 309 pacientes com EB hereditária, 278 foram incluídos. O tipo mais comum foi EB distrófica (EBD), com 73% (28,4% EBD dominante, 31,7% EBD recessiva e 12,9% EBD pruriginosa). Outros tipos foram juncional com 9,4%, simples com 16,5% e Kindler com 1,1%. Mulheres corresponderam a 53% e homens a 47%. História familiar foi encontrada em 35% e consanguinidade em 11%. A média de idade ao diagnóstico foi de 10,8 anos, e da idade atual foi de 26 anos. A média do tempo de seguimento foi de nove anos. Estenose de esôfago acometeu 14%, doenças dentárias acometeram 36%, desnutrição 13% e anemia 29% dos indivíduos. Durante a investigação diagnóstica, 72,6% realizaram exame histopatológico e 92% fizeram imunomapeamento. Nevos EB foram identificados em 17%. Nove pacientes tiveram CEC. Onze foram a óbito.
Limitações do estudoDados insuficientes nos prontuários, perda de seguimento e indisponibilidade do teste genético.
ConclusõesNeste estudo, predominou EB distrófica e ressaltou‐se a necessidade de cuidado multidisciplinar das comorbidades e das complicações.
Epidermólise bolhosa (EB) é um grupo de doenças hereditárias raras, até o momento incuráveis, geneticamente determinadas, caracterizadas por fragilidade da pele e mucosas. Há formação de bolhas mucocutâneas, erosões e ulcerações a pequenos traumas, o que causa impacto na qualidade de vida dos indivíduos acometidos.1
O consenso para classificação das EB de 20201 separou as formas clássicas de EB de outras doenças que cursam com fragilidade cutânea, em que a clivagem na pele é muito superficial (suprabasal), como síndrome da pele decídua.1 Nas formas clássicas de EB, o quadro clínico é heterogêneo. De acordo com o plano de clivagem na pele, as EB são divididas em quatro tipos principais: EB simples (EBS), EB juncional (EBJ), EB Ddistrófica (EBD) e EB Kindler (EBK).1 Mais de 30 subtipos clínicos são reconhecidos e foram descritas mutações patogênicas em 21 genes distintos, que codificam proteínas envolvidas na adesão e integridade celulares.2
Dados epidemiológicos no Brasil sobre EB são escassos.3,4 A associação DEBRA Brasil tem registro de 1.027 pacientes no país; entretanto, esse dado não está publicado. Os estudos brasileiros publicados são relatos e séries de casos com caracterização clínica e alguns com análise genética.3–13Em doenças raras, dados epidemiológicos confiáveis, com base em coortes bem caracterizadas, são essenciais.14 Ressaltam a necessidade de atenção ao cuidado e aos custos para os sistemas de saúde de um país específico. Também são importantes para o desenvolvimento e execução de estudos clínicos e para estimar o número de pacientes que poderiam se beneficiar com determinado tratamento.15 Estudos clínicos de fase 3 com Beremagene Geperpavec (B‐VEC) em pacientes com EBD mostraram resultados promissores quanto à cicatrização de feridas. Trata‐se de terapia tópica genética associada à edição gênica, com sequências de COL7A1 encapsuladas em vetores virais de herpes simples tipo 1.16
O objetivo desse estudo foi descrever os achados clínicos e epidemiológicos da EB hereditária de um hospital terciário em São Paulo, Brasil.
MétodosEstudo observacional, retrospectivo e transversal foi conduzido, após aprovação do Comitê de Ética da instituição, por meio da análise de prontuários físicos e eletrônicos de 309 pacientes diagnosticados com EB, na Divisão de Dermatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC‐FMUSP), Brasil, de 1 de janeiro de 2000 a 31 de dezembro de 2022.
O diagnóstico foi baseado na apresentação clínica, história familiar, comorbidades e nos exames histológico e de imunomapeamento obtidos por meio de material de biopsia de pele. Os diagnósticos foram agrupados nas seguintes categorias: EBS, EBJ, EBD dominante (EBDD), EBD recessiva (EBDR), EBD pruriginosa e EBK. Pacientes com dados incompletos ou prontuários médicos indisponíveis foram excluídos, assim como as doenças com clivagem suprabasal (consenso de 2020).1 Indivíduos com a doença e familiares também acometidos que não estavam registrados no nosso serviço não foram ativamente recrutados.
O banco de dados utilizado nesse estudo incluiu forma clínica de EB, sexo, história familiar de EB, presença de consanguinidade, idade de diagnóstico, idade atual, tempo de seguimento, comorbidades, realização de exame histológico e de imunomapeamento, presença de nevos EB e de tumores cutâneos, causa e idade de morte (se aplicável). Teste genético não foi incluído, pois não é ainda rotineiramente disponível no serviço. Dados estatísticos foram apresentados como porcentagens e médias.
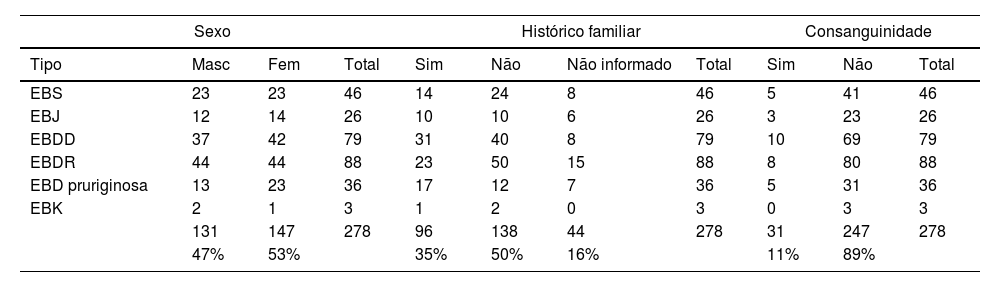
ResultadosNo total, dos 309 pacientes com EB, 31 foram excluídos, devido a dados incompletos. No total de 278 pacientes incluídos, 147 (53%) eram mulheres e 131 (47%), homens. O tipo mais comum foi a forma distrófica, com 203 pacientes (73%), dentre os quais 79 (28,4%) são EBDD (fig. 1A), 36 (12,9%) são EBD pruriginosa (fig. 1B) e 88 (31,7%) são EBDR (fig. 2). Outros tipos foram EBJ (fig. 3) com 26 (9,4%) pacientes, EBS (fig. 4) com 46 (16,5%) e tipo Kindler (fig. 5) em 3 (1,1%). História familiar positiva foi encontrada em 96 (35%) pacientes. Relato de consanguinidade esteve presente em 31 (11%) pacientes. A distribuição dos pacientes pelo tipo de EB em relação ao sexo, ao histórico familiar e à consanguinidade pode ser observada na tabela 1.
. (A) Forma generalizada acometendo o dorso. A involução das bolhas forma cicatrizes. (B) Pápulas escoriadas nos membros inferiores na EBD pruriginosa.")
. (A) Bolhas e áreas exulceradas ocasionam cicatrizes e fibrose. (B) Pseudosindactilia das mãos, com a fusão dos dígitos.")
. (A) Defeitos no esmalte dentário na arcada dentária inferior, com depressões puntiformes e coloração amarelada. Na arcada dentária superior é possível observar facetas laminadas (lentes de contato dentárias). (B) Fragilidade cutânea, áreas sem epitelização, outras áreas com tecido de granulação e crostas, no bebê.")
Epidermólise bolhosa juncional (EBJ). (A) Defeitos no esmalte dentário na arcada dentária inferior, com depressões puntiformes e coloração amarelada. Na arcada dentária superior é possível observar facetas laminadas (lentes de contato dentárias). (B) Fragilidade cutânea, áreas sem epitelização, outras áreas com tecido de granulação e crostas, no bebê.
. (A) Bolhas íntegras, exulcerações e crostas hemáticas na criança. Observar acometimento de áreas de fricção (região periaxilar). (B) Bolhas e erosões cicatrizando nos pés, na forma localizada de EBS.")
. (A) Lesões cicatriciais de disposição rendilhada, máculas hipo e hipercrômicas na criança com EBK. (B) Poiquilodermia na face. Crostas e exulcerações nos lábios. Dentes em mau estado de conservação.")
Distribuição do tipo de epidermólise bolhosa em relação ao sexo, ao histórico familiar e à consanguinidade
| Sexo | Histórico familiar | Consanguinidade | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tipo | Masc | Fem | Total | Sim | Não | Não informado | Total | Sim | Não | Total |
| EBS | 23 | 23 | 46 | 14 | 24 | 8 | 46 | 5 | 41 | 46 |
| EBJ | 12 | 14 | 26 | 10 | 10 | 6 | 26 | 3 | 23 | 26 |
| EBDD | 37 | 42 | 79 | 31 | 40 | 8 | 79 | 10 | 69 | 79 |
| EBDR | 44 | 44 | 88 | 23 | 50 | 15 | 88 | 8 | 80 | 88 |
| EBD pruriginosa | 13 | 23 | 36 | 17 | 12 | 7 | 36 | 5 | 31 | 36 |
| EBK | 2 | 1 | 3 | 1 | 2 | 0 | 3 | 0 | 3 | 3 |
| 131 | 147 | 278 | 96 | 138 | 44 | 278 | 31 | 247 | 278 | |
| 47% | 53% | 35% | 50% | 16% | 11% | 89% | ||||
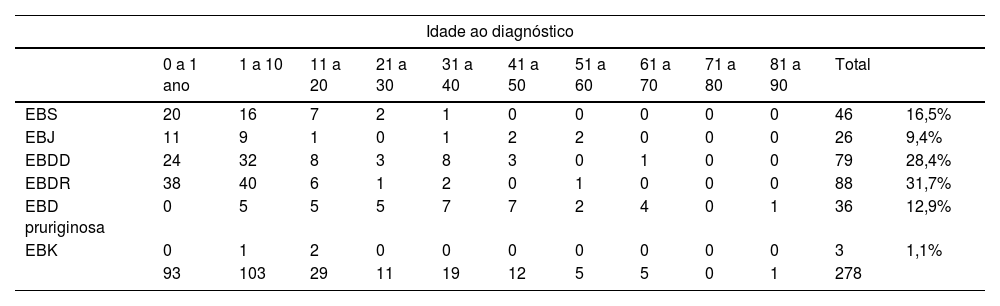
A média de idade ao diagnóstico foi de 10,8 anos (variou de 1 dia de vida até 82 anos, conforme detalhado na tabela 2). Noventa e três (33,5%) pacientes foram diagnosticados com menos de 1 ano de vida, enquanto 103 (37%) tinham entre 1 e 10 anos. Entre 11 e 20 anos de idade, 29 (10%) pacientes foram diagnosticados. Entre 21 e 80 anos, 52 (19%) pacientes obtiveram diagnóstico de EB, enquanto somente um paciente masculino tinha 82 anos.
Distribuição do tipo de epidermólise bolhosa quanto à idade ao diagnóstico e quanto à idade atual. No caso de pacientes que foram a óbito, foi considerada a idade da morte
| Idade ao diagnóstico | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 a 1 ano | 1 a 10 | 11 a 20 | 21 a 30 | 31 a 40 | 41 a 50 | 51 a 60 | 61 a 70 | 71 a 80 | 81 a 90 | Total | ||
| EBS | 20 | 16 | 7 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 46 | 16,5% |
| EBJ | 11 | 9 | 1 | 0 | 1 | 2 | 2 | 0 | 0 | 0 | 26 | 9,4% |
| EBDD | 24 | 32 | 8 | 3 | 8 | 3 | 0 | 1 | 0 | 0 | 79 | 28,4% |
| EBDR | 38 | 40 | 6 | 1 | 2 | 0 | 1 | 0 | 0 | 0 | 88 | 31,7% |
| EBD pruriginosa | 0 | 5 | 5 | 5 | 7 | 7 | 2 | 4 | 0 | 1 | 36 | 12,9% |
| EBK | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1,1% |
| 93 | 103 | 29 | 11 | 19 | 12 | 5 | 5 | 0 | 1 | 278 | ||
| Idade atual (ou do óbito) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 a 10 | 11 a 20 | 21 a 30 | 31 a 40 | 41 a 50 | 51 a 60 | 61 a 70 | 71 a 80 | 81 a 90 | Total | |||
| EBS | 8 | 14 | 15 | 5 | 3 | 1 | 0 | 0 | 0 | 46 | 16,5% | |
| EBJ | 6 | 6 | 9 | 0 | 0 | 1 | 3 | 1 | 0 | 26 | 9,4% | |
| EBDD | 13 | 28 | 15 | 8 | 4 | 7 | 3 | 0 | 1 | 79 | 28,4% | |
| EBDR | 13 | 41 | 25 | 6 | 0 | 2 | 1 | 0 | 0 | 88 | 31,7% | |
| EBD pruriginosa | 0 | 2 | 4 | 9 | 6 | 5 | 3 | 5 | 2 | 36 | 12,9% | |
| EBK | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1,1% | |
| 40 | 92 | 70 | 28 | 13 | 16 | 10 | 6 | 3 | 278 | |||
Em relação à idade atual (tabela 2), a média foi de 26 anos. Para o cálculo, a idade considerada no caso dos pacientes que foram à óbito foi a idade da morte. Somente 40 (14%) indivíduos tinham menos de 10 anos, enquanto 92 (33%) pacientes tinham entre 11 e 20 anos. Na faixa etária entre 21 e 80 anos, havia 143 (51%) pacientes, enquanto três tinham mais de 81 anos.
A média do tempo de seguimento foi de 9 anos, excluídos seis pacientes que compareceram somente para exame diagnóstico e não compareceram para seguimento no ambulatório. Para os pacientes falecidos, foi considerada a data da última consulta antes de o óbito acontecer.
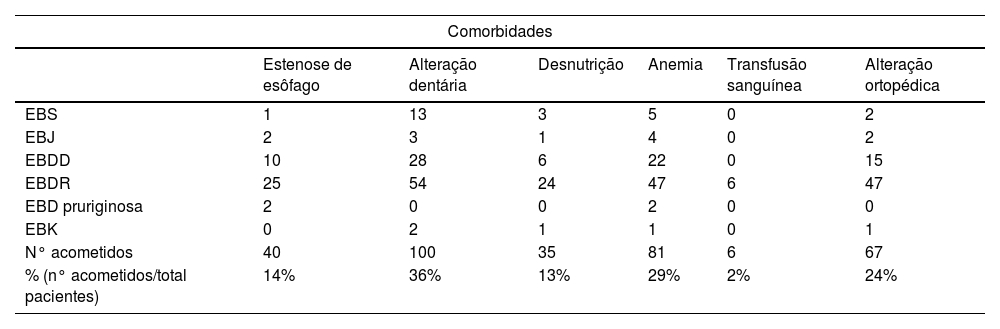
Em relação ao comprometimento extracutâneo (tabela 3), 40 (14%) pacientes tiveram diferentes graus de disfagia (estenose de esôfago). Desses, 25 (62,5%) são EBDR, 10 (25%) são EBDD, dois (5%) são EBD pruriginosa, dois (5%) são EBJ, um (2,5%) é EBS e nenhum é EBK.
Distribuição do tipo de epidermólise bolhosa em relação às principais comorbidades
| Comorbidades | ||||||
|---|---|---|---|---|---|---|
| Estenose de esôfago | Alteração dentária | Desnutrição | Anemia | Transfusão sanguínea | Alteração ortopédica | |
| EBS | 1 | 13 | 3 | 5 | 0 | 2 |
| EBJ | 2 | 3 | 1 | 4 | 0 | 2 |
| EBDD | 10 | 28 | 6 | 22 | 0 | 15 |
| EBDR | 25 | 54 | 24 | 47 | 6 | 47 |
| EBD pruriginosa | 2 | 0 | 0 | 2 | 0 | 0 |
| EBK | 0 | 2 | 1 | 1 | 0 | 1 |
| N° acometidos | 40 | 100 | 35 | 81 | 6 | 67 |
| % (n° acometidos/total pacientes) | 14% | 36% | 13% | 29% | 2% | 24% |
Doenças dentárias foram identificadas em 100 (36%) pacientes, dentre os quais 54 (54%) são EBDR, 28 (28%) são EBDD, 13 (13%) são EBS, três (3%) são EBJ, dois (2%) são EBK e nenhum é EBD pruriginosa.
Desnutrição foi avaliada por meio do índice de massa corporal (IMC ≤ 18) e acometeu 35 (13%) indivíduos. Desses, 24 (68,6%) são EBDR, seis (17,1%) são EBDD, três (8,5%) são EBS, um (2,9%) é EBJ e um (2,9%) é EBK.
Anemia foi achado frequente, com 81 (29%) pacientes acometidos; seis (2%) deles foram submetidos a transfusões sanguíneas, todos EBDR.
Acometimento ortopédico foi observado em 67 (24%) pacientes e aqueles com pseudosindactilia nas mãos (59 pacientes) foram encaminhados ao ambulatório de ortopedia.
Nevos da epidermólise bolhosa (nevos EB) foram identificados em 47 (17%) indivíduos, no total de 90 nevos, uma vez que um mesmo paciente apresentou de uma a cinco lesões (fig. 6A). Dez pacientes eram EBDD, 29 eram EBDR, dois eram EBJ, cinco eram EBS e um era EBD pruriginosa.
 Nevo da epidermólise bolhosa (nevo EB) no cotovelo, em paciente com EBDR. (B) Extensa área com exulceração e lesões de aspecto vegetante, com transformação para carcinoma espinocelular (CEC) no pé.")
Atresia de piloro foi identificada em uma paciente EBS com acometimento cutâneo grave. Distrofia muscular foi observada em um paciente EBS. Cardiomiopatia dilatada foi identificada em um paciente EBK de 30 anos que perdeu seguimento e em um paciente EBDR, que foi a óbito aos 20 anos.
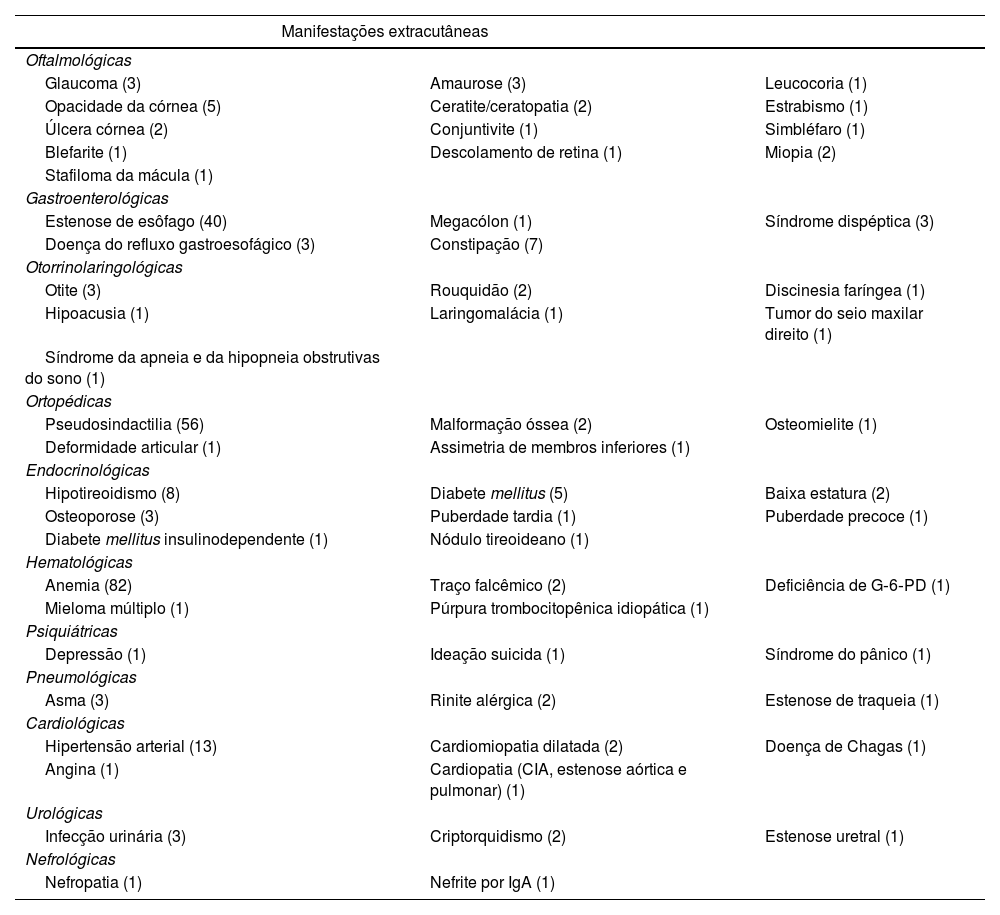
Alterações dermatológicas não relacionadas à EB incluem dermatite seborreica (1), dermatite atópica (1), pseudotínea amiantácea (1). Foliculite decalvante foi identificada em dois indivíduos, ambos com a forma distrófica de EB. Alopecia cicatricial acometeu duas pacientes EBJ. A tabela 4 mostra outras doenças extracutâneas encontradas nos pacientes dessa coorte.
Outras manifestações extracutâneas encontradas nos pacientes deste estudo, com o número de indivíduos acometidos entre parênteses
| Manifestações extracutâneas | ||
|---|---|---|
| Oftalmológicas | ||
| Glaucoma (3) | Amaurose (3) | Leucocoria (1) |
| Opacidade da córnea (5) | Ceratite/ceratopatia (2) | Estrabismo (1) |
| Úlcera córnea (2) | Conjuntivite (1) | Simbléfaro (1) |
| Blefarite (1) | Descolamento de retina (1) | Miopia (2) |
| Stafiloma da mácula (1) | ||
| Gastroenterológicas | ||
| Estenose de esôfago (40) | Megacólon (1) | Síndrome dispéptica (3) |
| Doença do refluxo gastroesofágico (3) | Constipação (7) | |
| Otorrinolaringológicas | ||
| Otite (3) | Rouquidão (2) | Discinesia faríngea (1) |
| Hipoacusia (1) | Laringomalácia (1) | Tumor do seio maxilar direito (1) |
| Síndrome da apneia e da hipopneia obstrutivas do sono (1) | ||
| Ortopédicas | ||
| Pseudosindactilia (56) | Malformação óssea (2) | Osteomielite (1) |
| Deformidade articular (1) | Assimetria de membros inferiores (1) | |
| Endocrinológicas | ||
| Hipotireoidismo (8) | Diabete mellitus (5) | Baixa estatura (2) |
| Osteoporose (3) | Puberdade tardia (1) | Puberdade precoce (1) |
| Diabete mellitus insulinodependente (1) | Nódulo tireoideano (1) | |
| Hematológicas | ||
| Anemia (82) | Traço falcêmico (2) | Deficiência de G‐6‐PD (1) |
| Mieloma múltiplo (1) | Púrpura trombocitopênica idiopática (1) | |
| Psiquiátricas | ||
| Depressão (1) | Ideação suicida (1) | Síndrome do pânico (1) |
| Pneumológicas | ||
| Asma (3) | Rinite alérgica (2) | Estenose de traqueia (1) |
| Cardiológicas | ||
| Hipertensão arterial (13) | Cardiomiopatia dilatada (2) | Doença de Chagas (1) |
| Angina (1) | Cardiopatia (CIA, estenose aórtica e pulmonar) (1) | |
| Urológicas | ||
| Infecção urinária (3) | Criptorquidismo (2) | Estenose uretral (1) |
| Nefrológicas | ||
| Nefropatia (1) | Nefrite por IgA (1) | |
No processo de investigação diagnóstica, 202 (72,6%) indivíduos foram submetidos ao total de 302 biopsias para exame histológico. Imunomapeamento foi realizado em 256 (92%) pacientes, com total de 359 exames realizados nesse período. Nove realizaram somente biopsia, 63 realizaram somente imunomapeamento, 193 pacientes foram submetidos a ambos os exames e 13 não fizeram nenhum. A classificação do tipo de EB dos 13 pacientes que não realizaram nenhum exame levou em consideração história familiar e quadro clínico compatível, apesar de o diagnóstico não ter sido confirmado laboratorialmente. Esses pacientes não foram submetidos à biopsia de pele ou imunomapeamento por questões sociais (indisponibilidade de retornar ao hospital) ou por falta de consentimento dos responsáveis ou dos próprios pacientes (cinco recém‐natos, cinco crianças menores de 12 anos e três adultos, idades 17, 27 e 49 anos).
Durante o período de seguimento, nove (3,2%) pacientes foram diagnosticados com 11 carcinomas espinocelulares (CEC). Um paciente EBJ apresentou três CEC (um em cada membro inferior e um no dorso). A localização do CEC (fig. 6B) nos demais pacientes foi nos membros inferiores em seis (um EBJ, quatro EBDR e um EBDD) e dorso em um paciente (EBDR). Em uma das pacientes EBDR, o local do CEC é desconhecido, por falta de informação nos registros médicos. Amputação transtibial foi realizada em três pacientes (dois homens EBDR aos 18 e aos 19 anos e uma mulher EBDD aos 27 anos). Cinco (55,5%) apresentaram metástase e foram a óbito, dentre os quais três EBDR (a idade do óbito de dois foi aos 18 anos e de um foi aos 20 anos), um EBDD (28 anos) e um EBJ (57 anos).
No total, 11 pacientes foram a óbito, cinco EBDR (18, 18, 20, 22 e 24 anos), cinco EBDD (1, 11, 15, 15, 28 anos) e um EBJ (57 anos). Causa de morte de cinco destes foi por complicações de CEC metastático. Três foram a óbito em virtude de sepse por broncopneumonia (dois EBDD, ambos com 15 anos, e um EBDR de 22 anos). Um paciente EBDR de 24 anos teve broncoaspiração como causa de morte. Duas crianças EBDD, de 1 ano e de 11 anos, foram a óbito sem causa conhecida.
Pacientes que não compareceram a consultas na dermatologia e em outras especialidades no HC‐FMUSP há mais de dois anos foram considerados perda de seguimento. No total, 158 (57%) pacientes tiveram data da última consulta antes de 31/12/2020. Entre 2021 e 2022, três pacientes foram a óbito.
DiscussãoEm nosso estudo de EB hereditária, não observamos predileção por sexo; o número de mulheres e homens acometidos foi semelhante, conforme previamente descrito na literatura.2,17 A forma distrófica de EB foi a mais frequente (73,4%), proporção semelhante à encontrada em estudo realizado no Irã (75,7%).17 Estudos de outros países descreveram frequência menor de EBD, como Canadá (35%),18 Austrália (35%),19 Holanda20 com 34,7% (EBDD 23,1% e EBDR 11,4%), Alemanha (32,5%)21 e Índia (17%).22 A predominância de formas mais graves de EB em nosso estudo se deve, em parte, a dificuldades de acesso a atendimento médico especializado23 em nosso país e subdiagnóstico das formas mais localizadas de EBS e ao fato de o HC‐FMUSP ser hospital terciário, referência nacional no atendimento de paciente com EB, e atender casos de mais alta complexidade.
No presente estudo, EBS acometeu 16,5% dos pacientes, enquanto EBJ 9,4% dos pacientes e EBK 1,1%. A frequência de EBJ no estudo do Irã,17 Canadá18 e Austrália19 foi de 11%; na Holanda20 de 18,8% e na Alemanha,21 17,1%. Já EBK, 3% no Irã,17 0,3% na Austrália,19 0,9% na Holanda20 e 0,5% na Alemanha.21 Em ambos os tipos de EB, as porcentagens encontradas em nosso estudo foram semelhantes. Em relação à EBS, a frequência encontrada é menor do que a descrita na literatura, uma vez que a população estudada advém de hospital terciário. Segundo estudo epidemiológico de 2021da Holanda,20 EBS foi diagnosticada em 45,7%. Nessa coorte holandesa, 90,5% dos pacientes tiveram diagnóstico geneticamente confirmado.20 Dados epidemiológicos de 1.779 pacientes da Alemanha, publicados em 2022,21 mostraram 39,3% (700) de pacientes EBS. Este estudo incluiu pacientes de hospitais terciários, ambulatórios de Dermatologia, laboratórios que realizaram o diagnóstico e de organizações de pacientes.
A média da idade atual encontrada em nosso estudo foi de 26 anos, e somente 40 (14%) indivíduos tinham menos de 10 anos. O elevado número de pacientes na idade puberal, ou seja, acima de 11 anos, permite inferir aumento na expectativa de vida e reflete a melhora nos cuidados multidisciplinares, principalmente em relação à nutrição, infecção e manejo da dor.
Quanto à consanguinidade, apenas 31 pacientes (11%) declararam sua ocorrênci, e a EBDR foi o tipo que mais acometeu a população em estudo (25,8%). Como a maioria dos pacientes não realizou teste genético, o diagnóstico não foi confirmado por esse método. Na literatura, é relatado que o número de pacientes com forma autossômica recessiva de EB pode estar associado às comunidades com altas taxas de consanguinidade parental.24
As queixas mais comuns dos pacientes foram alterações dentárias (36%), seguido por anemia (29%) e doenças ortopédicas (24%). No estudo do Irã, foi disfagia seguido de alterações dentárias.17 Esses achados dentários se devem na EBJ à hipoplasia do esmalte dentário e presença de depressões puntiformes (fig. 3A), que são achados clínicos patognomônicos nessa forma de EB. Essas alterações acarretam perda precoce de dentes e cáries. Na EBDR, microstomia e anquiloglossia são observadas e dificultam a higiene dental diária.25 Deve‐se sempre levar em consideração os cuidados precários de higiene bucal na maioria dos doentes com EB (fig. 5B).
Pseudosindactilia das mãos (fig. 2B) e dos pés é complicação músculo‐esquelética que ocorre principalmente na EBDR.25 O uso de luvas ou curativos para manter os quirodáctilos separados é aconselhado nos pacientes com EB distrófica. Em 2011, o grupo de ortopedia especializado em mãos do HC‐FMUSP, em conjunto com a equipe de Dermatologia pediátrica, publicou dados de 59 pacientes com EB e deformidades nas mãos, dos quais 25 foram submetidos à cirurgia, com taxa elevada de reoperação.26 Estudo de 2019 do grupo da China27 relatou 11 pacientes EBDR submetidos à cirurgia e com seguimento por dois anos, dos quais nove apresentaram perda da funcionalidade das mãos pelo reestreitamento dos espaços interdigitais, com adesão de dígitos e flexão de articulações metacarpofalangeanas e interfalangeanas. Os outros dois pacientes foram reoperados após um ano em virtude de recidiva.
O Registro Nacional de EB (NEBR) dos Estados Unidos com casuística de 3.280 pacientes acompanhados por período de 16 anos (1986‐2002)28,29 permanece como o maior estudo epidemiológico sobre EB. Demonstrou que a frequência de estenose de esôfago é de 79,1% em pacientes com EBDR grave, 37,2% para EBDR intermediária e 14,3% em EBJ, com frequências menores nos demais subtipos de EB.28 Em nosso estudo, estenose de esôfago foi identificada em 14% do total de indivíduos; 28,4% e 7,7% foram as frequências observadas em pacientes com EBDR e EBJ, respectivamente.
Em 2005, foi publicado um trabalho envolvendo a Gastroenterologia e a Dermatologia Pediátrica do HC‐FMUSP,30 que relatou 19 pacientes com formas graves de EB (EBD e EBJ) atendidos entre janeiro de 1999 e abril de 2001. Vômitos e disfagia foram referidos por 16 (84,2%) casos, engasgos por 14 (73,7%) e constipação intestinal por 14 (73,7%) pacientes. Anemia acometeu todos os casos. Em relação aos aspectos nutricionais, 12 (63,1%) apresentavam peso e estatura inferiores ao percentil 2,5.30 No presente estudo, desnutrição acometeu 35 (12%). O menor percentual encontrado está associado à metodologia deste trabalho, que incluiu todas as formas de EB.
Nevo EB é descrito como nevo adquirido, eruptivo, assimétrico e acomete cerca de 14% dos portadores de EB, em suas várias formas clínicas.31 Os nevos EB geralmente ocorrem em sítios de bolhas prévias e podem involuir espontaneamente. Entretanto, teoricamente o melanoma poderia surgir a partir do nevo EB ou de lesões pigmentadas que mimetizam clinicamente o nevo EB. Dessa maneira, qualquer mudança morfológica no nevo EB, em especial o surgimento de nodulação ou ulceração, aponta para a realização de estudo histopatológico.32 Neste estudo, nevos EB foram identificados em 17% dos pacientes. Em 2014, a equipe de Dermatologia Pediátrica do HC‐FMUSP descreveu 13 pacientes EBD com nevos EB, cinco dos quais foram biopsiados em virtude das características dermatoscópicas atípicas dos nevos. Dois corresponderam a nevo atípico ou proliferação lentiginosa atípica à avaliação histopatológica.7 Em 2005, um grupo da Áustria33 descreveu características dermatoscópicas de 23 nevos EB de 11 pacientes, com critérios associados a melanoma encontrados, como padrão multicomponentes (20 de 23), rede pigmentada atípica (17 de 23), glóbulos e pontos irregulares (16 de 23), pigmentação irregular (22 de 23) e padrão vascular atípico (7 de 23). O mesmo grupo indicou avaliação histológica de lesões altamente suspeitas.
A atresia de piloro associada à EB foi encontrada em uma paciente feminina EBS de 28 anos e com acometimento cutâneo extenso. Trata‐se de um subtipo distinto, que geralmente ocorre em fenótipos graves de EB.34 Quando ocorre concomitantemente à EBS grave, está associada à mutação no gene da plectina (PLEC), proteína formadora do hemidesmossoma.2 O diagnóstico genético da paciente em questão ainda não foi realizado.
Um paciente masculino EBS de 22 anos com diagnóstico genético de mutação no gene PLEC apresentou distrofia muscular, como já citado na literatura. Pode estar presente ao nascimento ou nos primeiros dias de vida nas crianças com doença cutânea grave,35 ou pode se estabelecer de forma insidiosa no final da infância ou na vida adulta.25 No estudo holandês de 2021,20 dois pacientes com EBS e distrofia muscular morreram com idades de 43,7 anos e 46 anos por falência cardíaca.
O maior risco de cardiomiopatia dilatada é visto entre pacientes EBDR grave, com risco cumulativo de 4,51% aos 20 anos de idade.36 Dentre os pacientes incluídos neste estudo, cardiomiopatia acometeu um paciente EBDR, que foi a óbito aos 20 anos, e um paciente EBK de 30 anos, que perdeu seguimento.
Durante o período avaliado, foram realizadas 302 biopsias de pele e 359 imunomapeamentos. Alguns exames foram repetidos em virtude de laudos inconclusivos, pois as bolhas biopsiadas estavam em processo de reepitelização ou a clivagem estava ausente. O exame histológico da biopsia de pele deve ser realizado em uma bolha íntegra (pele acometida) recente. A clivagem é intraepidérmica nos casos de EBS e subepidérmica na EBJ e EBD, não sendo possível diferenciar essas duas formas por meio da histopatologia com coloração de rotina.10 Na EBK, o plano de clivagem é variável. Já o imunomapeamento pode ser realizado em amostra de pele sã com vesícula induzida ou em vesícula recente e idealmente pequena, para que não se rompa no ato da biopsia. Trata‐se de técnica de imunofluorescência e tem a vantagem de possibilitar a visualização do plano de clivagem, permitindo diferenciação entre as formas simples, juncional e distrófica.10 Os marcadores utilizados no Departamento de Dermatologia do HC‐FMUSP são: antígeno do penfigoide bolhoso (marcador de hemidesmossoma), anticorpo antilaminina 5 (lâmina lúcida), anticorpo anticolágeno IV (lâmina densa) e anticorpo anticolágeno VII (sublâmina densa e fibrilas de ancoragem). Na EBS, a clivagem ocorre na camada basal. Na EBJ, o nível de clivagem ocorre na lâmina lúcida, enquanto na EBD ocorre na sublâmina densa (fig. 7).10 Na EBDR, há ausência ou diminuição acentuada da fluorescência com o colágeno VII. Em 2021, um estudo da Índia37 mostrou a utilidade do imunomapeamento como método diagnóstico, frente à indisponibilidade de microscopia eletrônica e de testes genéticos. Diagnóstico de 104 EBS, 28 EBJ e de 26 EBD foi realizado com taxa de concordância de 41,3%, com maiores taxas nos casos de EBS.
. Observar clivagem na sublâmina densa, com depósito dos marcadores e fluorescência no teto da bolha. Os marcadores utilizados são: (A) Antígeno do penfigoide bolhoso, (B) laminina, (C) colágeno IV e (D) colágeno VII.")
A pesquisa das mutações nos genes associados à EB é padrão ouro para definição do subtipo de EB e evitaria a realização de biopsia e imunomapeamento. Tem sido realizada de maneira mais rápida e menos custosa, com maiores taxas de detecção mutacional, devido aos avanços na tecnologia de sequenciamento.38 No nosso serviço, teste genético vem sendo realizado no âmbito da pesquisa, porém ainda não está amplamente disponível na prática ambulatorial por não ser exame coberto pelo SUS.
Em algumas formas de EB, especialmente na EBDR, há risco aumentado de CEC no final da adolescência e início da vida adulta. Estudo recente do grupo da Austrália e da Nova Zelândia39 mostrou 16 pacientes EBDR com 161 CEC primários, com média de 10 CEC por indivíduo. Desses tumores, 70% surgiram nas mãos e nos pés, principalmente em áreas de úlceras crônicas e de feridas que não cicatrizavam. Metástase foi detectada em 11 (68,7%) desses pacientes. Amputação parcial do membro foi realizado em sete casos.39 Dos nove pacientes diagnosticados com CEC em nosso estudo, sete (77,7%) apresentaram tumor nos membros inferiores e cinco (55,5%) tiveram metástase. Apenas um indivíduo EBJ apresentou mais de um CEC (três tumores), enquanto três pacientes foram submetidos à amputação transtibial. CEC é a principal causa de morte nesse grupo de pacientes, e o risco cumulativo de morte por metástase é de 38,7% aos 35 anos e de 78,8% aos 55 anos de idade. A maioria dos pacientes com EBDR morrem de CEC metastático no período de cinco anos após o diagnóstico.29
O baixo número de óbitos (11) identificado neste estudo pode estar relacionado aos cuidados multidisciplinares dirigidos aos pacientes. Porém, há o viés de informação, pois esse dado somente é registrado no prontuário quando o óbito ocorre em algum hospital do complexo HC‐FMUSP. Informações sobre óbitos que ocorrem em casa ou em outros hospitais não estão disponíveis, pela falta de um banco de dados unificado nacional. Cinco pacientes EBDR foram a óbito com média de idade de 20,4 anos, enquanto a média de idade de óbito de cinco pacientes EBDD foi de 14 anos. A idade média de vida em pacientes com EBD segundo estudo recente na Holanda é de 32,5 anos. Em EBDR, esse valor é de 28,8 anos e em EBDD de 33,3 anos.20
Quanto às principais limitações, por se tratar de um estudo retrospectivo com longo período de avaliação (2000‐2022), houve falta de informações em alguns prontuários médicos e perda de seguimento de pacientes. Outro ponto é a indisponibilidade de testes genéticos de rotina para os pacientes com EB em nosso serviço e no sistema público de saúde, o que pode dificultar a classificação da forma clínica de EB. Apesar desse estudo não ser multicêntrico, representa o primeiro trabalho com dados epidemiológicos de epidermólise bolhosa hereditária na população brasileira.
No Brasil, o HC‐FMUSP é um dos poucos centros de referência multidisciplinar especializados em EB. O acesso a esses serviços escassos é difícil, uma vez que depende do encaminhamento das unidades básicas de saúde, que muitas vezes são distantes e localizam‐se até mesmo em outros Estados, de forma que o nosso serviço atende pacientes de todo o país. Outro problema crítico é a falta de suprimento de necessidades básicas dos pacientes, como curativos não‐aderentes e até mesmo curativos simples. Muitas vezes o paciente de EB não é diagnosticado e tratado adequadamente. Há subdiagnóstico e os poucos dados epidemiológicos que são publicados acabam não refletindo a realidade da incidência e prevalência de doenças raras, como a EB. Semelhante ao que ocorre no Canadá, não há centros especializados no cuidado de adultos com EB e esses continuam sendo atendidos em ambulatórios pediátricos. Questões da vida adulta, como sexualidade, emprego, independência e manejo de câncer de pele são tratados com limitações, o que torna necessário o encaminhamento a outras especialidades.40
ConclusãoEm doenças raras como a EB, dados epidemiológicos confiáveis permitem a identificação de tendências epidemiológicas.14 Esse estudo demonstra a predominância de formas mais graves na nossa casuística (EB distrófica), provavelmente por se tratar de um hospital terciário, e ressalta a necessidade de cuidado multidisciplinar, principalmente em relação às comorbidades e às complicações dos pacientes, como anemia, doenças dentárias, ortopédicas e do trato gastrointestinal e carcinoma espinocelular. No Brasil, há a necessidade de criação de banco de dados que unifique informações clínicas de toda a população, a fim de identificar essas tendências e permitir melhor direcionamento das políticas de saúde (verbas e financiamento).
Suporte financeiroEstudo patrocinado pela Fundo de Apoio à Dermatologia do Estado de São Paulo (FUNADERSP) [PROJETO 105‐2022] e pelo Departamento de Dermatologia do Hospital das Clínicas da Universidade de São Paulo.
Contribuição dos autoresChan I. Thien: concepção e planejamento do estudo; coleta de dados ou análise e interpretação dos dados; análise estatística; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; aprovação da versão final do manuscrito.
Vanessa Rolim Bessa: Coleta de dados, ou análise e interpretação dos dados; análise estatística; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Isadora Zago Miotto: Coleta de dados, ou análise e interpretação dos dados; análise estatística; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Luciana Paula Samorano: Concepção e planejamento do estudo; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Maria Cecília Rivitti Machado: Concepção e planejamento do estudo; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Zilda Najjar Prado de Oliveira: Concepção e planejamento do estudo; elaboração e redação do manuscrito ou revisão crítica de conteúdo intelectual importante; análise estatística; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Ao dr. Marcello Menta Simonsen Nico, professor do Departamento de Dermatologia do HCFMUSP e chefe do ambulatório de Estomatologia do Hospital HC‐FMUSP, que cedeu fotos clínicas para este artigo.
Ao dr. Alexandre Marques Périgo, biólogo do Laboratório de Imunopatologia da Pele, do Departamento de Dermatologia do Hospital HC‐FMUSP, que cedeu fotos de imunomapeamento para este artigo.
Como citar este artigo: Thien CI, Bessa VR, Miotto IZ, Samorano LP, Rivitti‐Machado MC, Oliveira ZN. Hereditary epidermolysis bullosa: clinical‐epidemiological profile of 278 patients at a tertiary hospital in São Paulo, Brazil. An Bras Dermatol. 2024;99:380–90.
Trabalho realizado no Departamento de Dermatologia, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brazil.
