

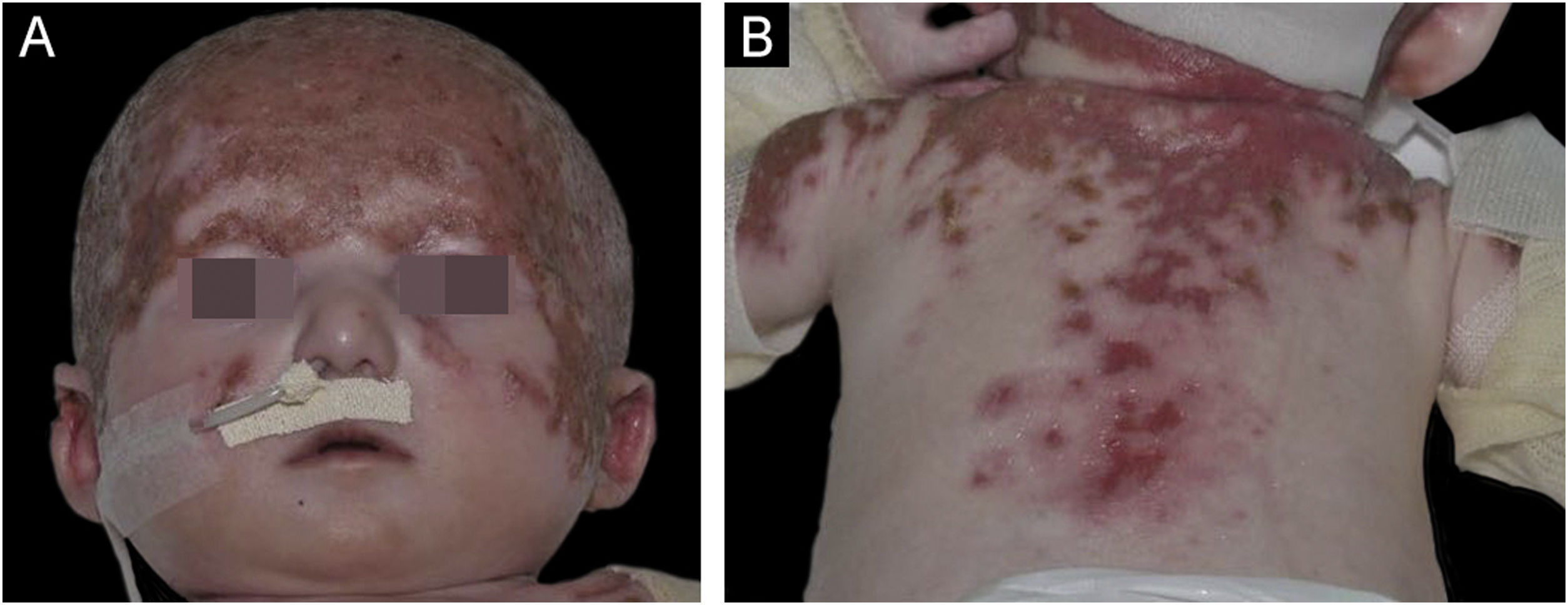
O presente relato discute a evolução e condução de um caso de síndrome de Hay‐Wells (SHW), previamente publicado nesta revista, de paciente do sexo masculino, atualmente com 9 anos, acompanhado desde os 4 meses de vida.1 Com pais não consanguíneos e irmãos hígidos, nascido a termo e por parto vaginal, apresentava desde o nascimento lesões exulceradas no tronco, couro cabeludo, face e membros superiores (fig. 1); onicodistrofia (fig. 2); anquiloblefaron filiforme adnatum – corrigida na sala de parto; fenda palatina; deformidade do pavilhão auricular; e micropênis.1 O paciente ficou internado durante os primeiros três meses de vida por perdas transepidérmicas com distúrbios hidroeletrolíticos, infecções cutâneas, otite e pneumonia. Foi encaminhado inicialmente com diagnóstico de epidermólise bolhosa; entretanto, os diversos achados clínicos permitiram o diagnóstico de SHW.
 Exulcerações no couro cabeludo e face. (B) Exulcerações no dorso e região proximal dos membros superiores.")
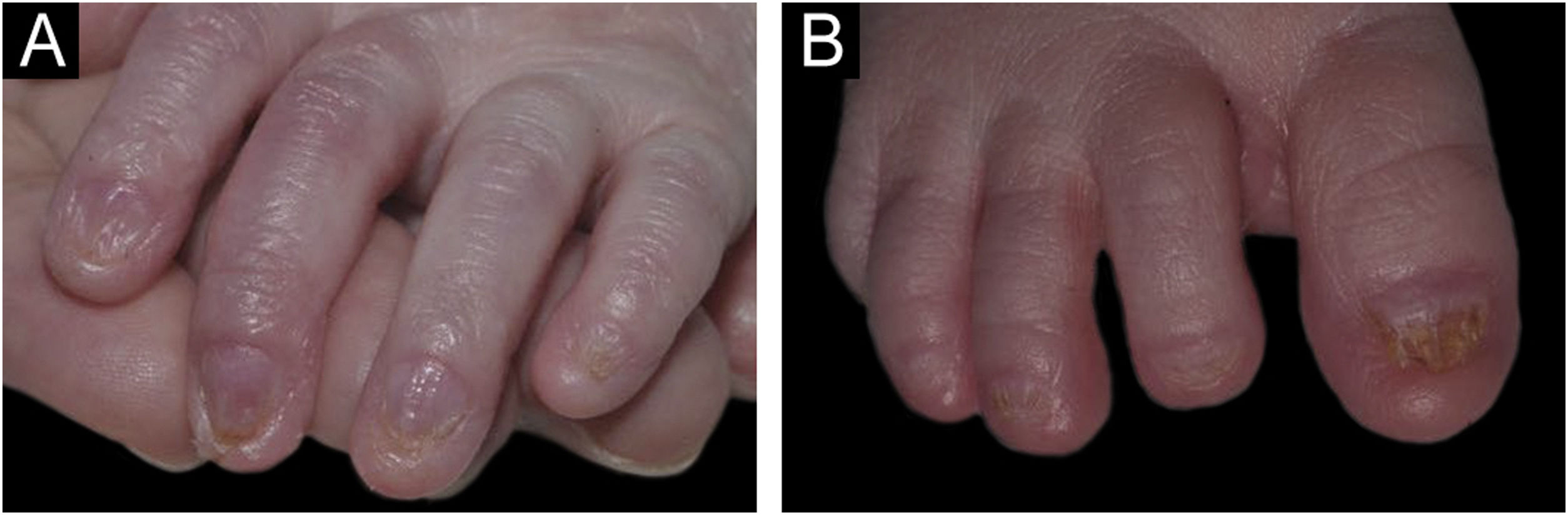
 Presença de onicodistrofia em todos os quirodáctilos. (B) Presença de onicodistrofia em todos os pododáctilos.")
Desde o nascimento, o paciente evoluiu com intolerância e recusa de dieta por via oral (VO), necessitou de sonda nasoenteral, que foi substituída por gastrostomia aos 3 anos, momento em que também realizou a correção da fenda palatina. Aos 4 anos, o paciente foi diagnosticado com fimose e aderências balanoprepuciais, já corrigidas, e hipospadia glandar, que não exigiu intervenção. O paciente iniciou melhora da tolerância à alimentação VO apenas aos 7 anos.
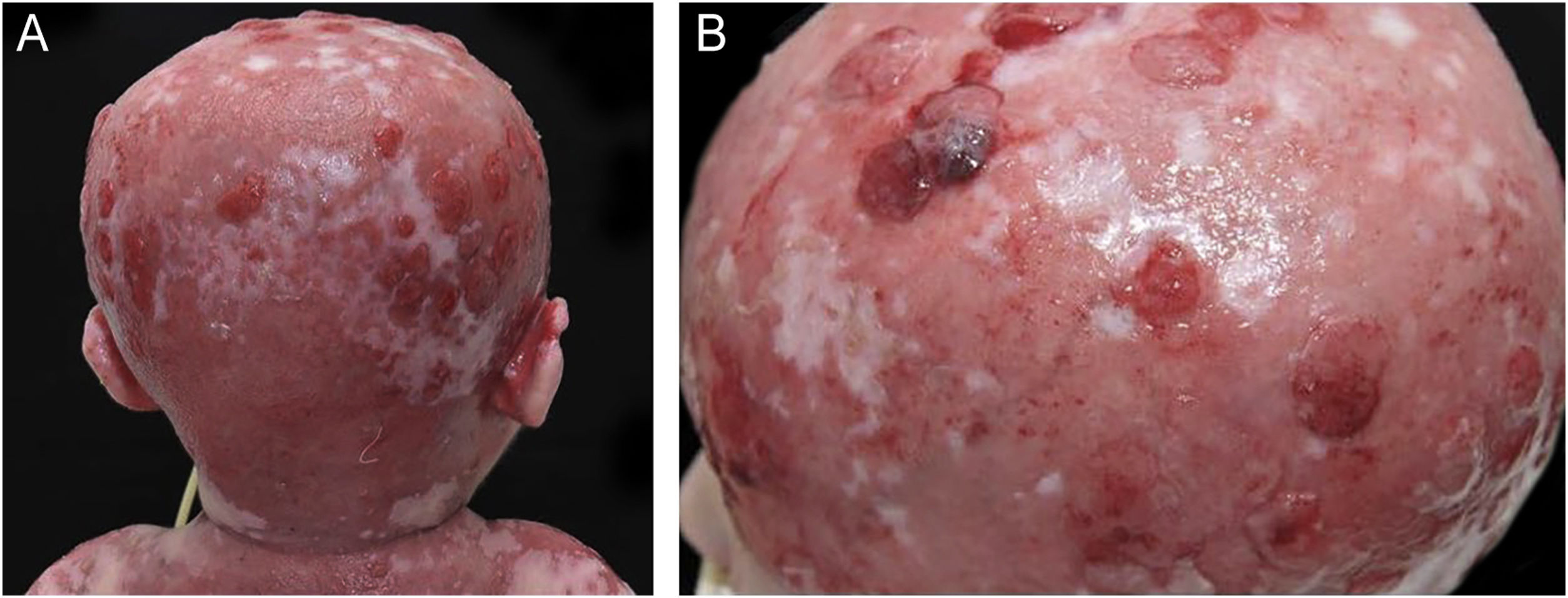
As exulcerações cutâneas, principalmente as do couro cabeludo, tiveram diversos episódios de tecido de granulação exuberante e colonização bacteriana desde o nascimento, sem melhora nos primeiros anos de vida (fig. 3), a despeito dos cuidados locais e das terapias antimicrobianas tópicas e sistêmicas. Foram realizadas diversas culturas do exsudato dessas lesões, com resultados positivos para Staphylococcus aureus resistentes à meticilina com perfil comunitário (CA‐MRSA) e Pseudomonas aeruginosa.
 e região parietal (B).")
Os cuidados cutâneos iniciais foram centrados na remoção delicada de escamas e crostas com uso de curativos não aderentes. Para os tecidos de granulação exuberante, foram realizadas diversas sessões de desbridamento cirúrgico superficial, com anestesia tópica e uso de curativos específicos, que absorvessem exsudato, preferencialmente com prata, para controle e resolução da colonização bacteriana local. Inicialmente, optou‐se por curativos de espuma com prata (hidropolímeros), por serem recortados no formato necessário, pelo bom poder de absorção de exsudato e perda de umidade por evaporação, mantendo o isolamento térmico e ambiente úmido e com trocas a cada quatro dias, objetivando melhorar a qualidade de vida do paciente, aliviar a dor, acelerar a cicatrização e prevenir episódios de colonização e infecção.
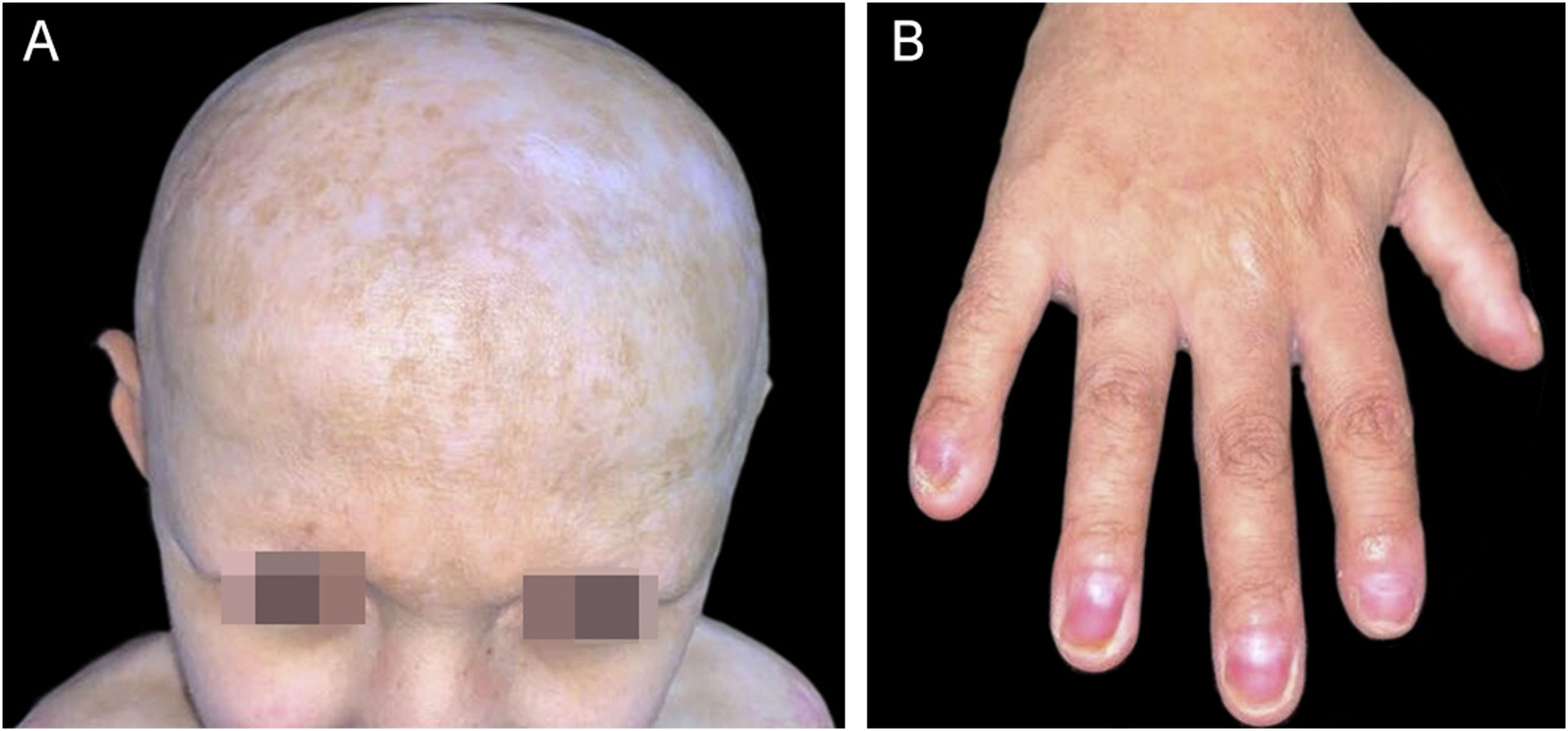
Desde os 7 anos o paciente não apresentou novas exulcerações e não teve mais necessidade de uso de tratamentos específicos. Ao exame dermatológico atual, são identificados deformidade do pavilhão auricular, micropênis, alopecia, madarose e onicodistrofia, além de hipodontia, dentes cônicos, cicatrizes hipo/hipercrômicas, principalmente na face, região cervical, couro cabeludo e tronco anterior; e cicatrizes hipertróficas nos membros superiores, tronco anterior e dorso (fig. 4). O paciente relatou anidrose generalizada e intolerância ao calor, evidenciada ao exame como xerose generalizada, sem descamação.
 Enfoque em alopecia cicatricial. (B) Onicodistrofia em todos os quirodáctilos e hiponíquia visualizada em 1° e 5° quirodáctilos.")
O paciente faz acompanhamento atual com neuropediatria (atraso leve no neurodesenvolvimento psicomotor), fonoaudiologia (dificuldade de fala), gastropediatria, urologia, otorrinolaringologia (membrana timpânica com perfuração puntiforme decorrente de otites recorrentes), oftalmologia (em uso de lágrimas artificiais), nutrição (baixa estatura) e dermatologia.
Diversas síndromes autossômicas dominantes resultam da mutação do gene p63, tais como a síndrome EEC (ectrodactyly, ectodermal dysplasia, clefting), a síndrome ADULT (acro‐dermato‐ungual‐uacrimal‐tooth), síndrome membro‐mamária, a SHFM (split‐hand/foot malformation) e a SHW ou AEC (ankyloblepharon, ectodermal dysplasia and cleft lip/palate syndrome). Essas síndromes cursam com displasia ectodérmica, alterações dos membros e fissura orofacial. Os fenótipos diferentes se justificam por diferentes mutações que podem ocorrer no mesmo gene, seja com ganho ou perda de função. O gene p63 codifica seis proteínas diferentes que terão seus efeitos funcionais alterados de variadas formas a depender da síndrome em questão.2–6 Aproximadamente 30% dos pacientes apresentam um progenitor afetado, enquanto 70% dos casos surgem de mutação de novo.5
A SHW/AEC compartilha de algumas características comuns com síndrome EEC. Entretanto, o envolvimento ectodérmico é muito mais grave na síndrome SHW/AEC, na qual ocorrem mais frequentemente lesões do couro cabeludo e de modo mais grave. Além disso, a fissura facial, quando presente na síndrome EEC, é sempre fissura labial (com ou sem fissura palatina), enquanto apenas fissura palatina ocorre na SHW. Envolvimento de membros na AEC é mínima ou ausente.6
As exulcerações na SHW são comuns ao nascimento, com risco de levar a alopecia cicatricial e hipotricose. Cerca de 70% dos neonatos apresentam anquiloblefaron filiforme adnatum com severidade variável. Também pode existir agenesia ou atresia dos pontos lacrimais, levando a conjuntivite ou blefarite crônica. A maioria apresenta distrofia ungueal e anormalidades dentárias, como hipodontia e dentes cônicos. A sudorese pode ser diminuída, gerando intolerância térmica. Fissura palatina com ou sem lábio leporino ocorre em todos os casos. Alterações na posição da língua e dificuldade de fala também já foram descritas.5,7–9 Na SHW, devem ser garantidos os cuidados intensivos necessários e posterior acompanhamento individualizado, de acordo com as demandas exibidas ao longo do desenvolvimento infantil e na fase adulta, realizando cuidado continuado e multidisciplinar. De fato, após o período da primeira infância – quando o risco de complicações graves como infecções e perda de líquidos é maior –, o prognóstico geral é bom, com expectativa de vida normal, como evidenciado neste caso, com melhora significante das lesões cutâneas após os 7 anos de vida.5,9
Suporte financeiroNenhum.
Contribuição dos autoresRebecca Perez de Amorim: Concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Maria Vitória Yuka Messias Nakata: Elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Vitória Gabrielle De Paula Gonçalves: Elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Ivanka Miranda de Castro: Obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Gabriela Roncada Haddad: Concepção e planejamento do estudo; participação efetiva na orientação da pesquisa; revisão crítica da literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Luciana Patrícia Fernandes Abbade: Concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Amorim RP, Nakata MVYM, Gonçalves VGP, Castro IM, Haddad GR, Abbade LPF. Hay‐Wells Syndrome: the challenges of a nine‐year follow‐up. An Bras Dermatol. 2024;99:964–6.
Trabalho realizado na Faculdade de Medicina, Universidade Estadual Paulista, Botucatu, SP, Brasil.
