







Purpura is defined as a visible hemorrhage in the skin or mucosa, which is not evanescent upon pressure. Proper classification allows a better patient approach due to its multiple diagnoses. Purpuras can be categorized by size, morphology, and other characteristics. The course varies according to the etiology, as do the diagnostic approach and treatment. This review discusses pigmented purpuras and some cutaneous vascular occlusion syndromes.
Purpura is defined as a visible hemorrhage in the skin or mucosa that is not evanescent upon pressure. Proper classification provides a better patient approach due to the multiple diagnoses of purpura. Purpuras can be classified by size, morphology, pathophysiology, and other characteristics (Table 1).
Purpura classification
| Size | Morphology | Characteristics |
|---|---|---|
| ≤ 4mm = petechiae | Retiform | Inflammatory |
| 5 to 9mm = macule | Non-retiform | Non-inflammatory |
| ≥ 1cm = ecchymosis | Livedo reticularis | |
| Livedo racemosa |
Source: Piette WW, 2012.1
Regarding morphology, retiform purpura should be differentiated from livedo reticularis and livedo racemosa. These conditions have a similar morphological appearance, characterized as violaceous macules in a net-like, arborized, or starry form but they differ in pathophysiology (Chart 1).
Livedo reticularis, livedo racemosa, and retiform purpura characteristics
| Pathophysiology | Topography | |
|---|---|---|
| Livedo reticularis | Low blood flow due to low output state. | Lower extremities |
| Livedo racemosa | Irregular blood flow due to mechanical obstruction. | Trunk and lower extremities |
| Retiform purpura | Purpura and necrosis due to venous occlusion. | Variable |
Source: Wysong A, et al, 2011.16
The clinical course varies according to the etiology. Macular purpura or non-palpable purpura heals faster and exhibits a color transition from red-blue to violaceous, green, yellow, or brown due to extravasation of erythrocytes and few inflammatory cells.1 Palpable purpura takes longer to heal due to the presence of inflammatory cells and immune complex deposits that lead to vascular occlusion.1
Purpura has a long list of differential diagnoses, which differ particularly in their pathophysiology. One way to approach purpura is by answering the question, “Is the patient bleeding?”, since some bleeding disorders require urgent treatment (Figure 1). The purpose of this review is to describe some of the differential diagnoses and their physiopathogenic mechanisms to provide a better patient approach and guide treatment. This article will discuss pigmented purpuras and some cutaneous vascular occlusion syndromes. Although vasculitis is part of the differential diagnoses, it will not be discussed in this review.

Purpura: diagnostic algorithm
NPH: nocturnal paroxysmal hemoglobinuria; TTP: thrombotic thrombocytopenic purpura; APS: antiphospholipid syndrome; HIT: heparin-induced thrombocytopenia; ITP: ldiopathic thrombocytopenic purpura; NSAID: non-steroidal anti-inflammatory drugs; DIC: disseminated intravascular coagulation
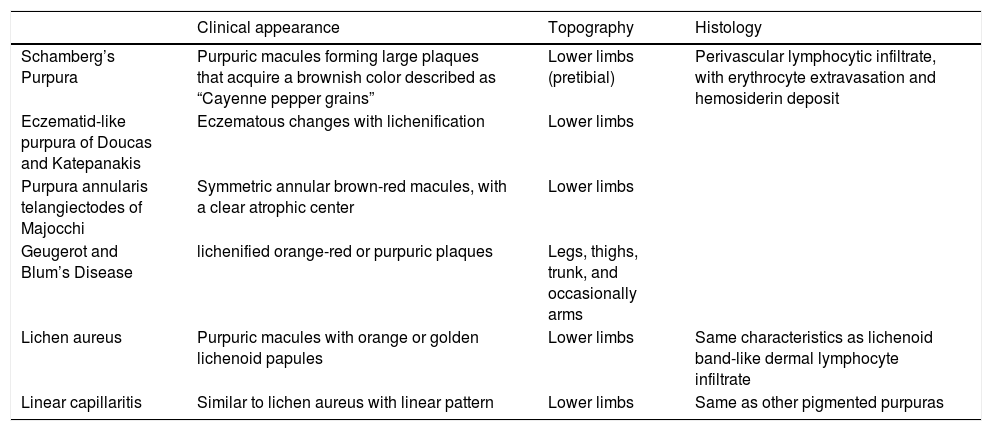
Pigmented purpuras, also known as chronic pigmented purpuric dermatosis, purpura simplex, and capillaritis, among others, encompasses five major clinical variants, including Schamberg’s purpura, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, eczematid-like purpura of Doucas and Kapetanakis, and lichen aureus.1-4 Each condition is associated with different triggers, which are difficult to establish in practice and do not appear to have therapeutic or prognostic implications.3,4
Capillaritis is associated with many triggers, including venous hypertension, exercise, pregnancy, frail capillaries, drug toxicity from acetaminophen, aspirin, hydralazine, and thiamine, and hyperactive substances such as textiles, colorants, and alcohol.2,3 Idiopathic capillaritis is the most common form, since most cases are not associated with a specific trigger.4
Epidemiological data are lacking. However, a 5-fold increased prevalence is noted in men compared to women. The condition predominantly affects adults 40 to 60 years of age.2,5 Some variants predominate in children and young adults.3-5
The physiopathogenesis is unknown, but the condition is believed to be due to a cutaneous hypersensitivity reaction that causes capillary fragility and permeability, leading to erythrocyte extravasation and hemosiderin deposits noted on biopsy.1,2,6 The following three main pathogenic theories have been described according to histological data: vascular fragility, humoral immunity, and cellular immunity.2,3
Pigmented purpuras present as petechiae or pigmented macules on distal lower extremities.1-4 These lesions can be generalized, due to self-limiting viral infection, or localized. All variants have specific distinguishing clinical characteristics (chart2).
Pigmented purpuras
| Clinical appearance | Topography | Histology | |
|---|---|---|---|
| Schamberg’s Purpura | Purpuric macules forming large plaques that acquire a brownish color described as “Cayenne pepper grains” | Lower limbs (pretibial) | Perivascular lymphocytic infiltrate, with erythrocyte extravasation and hemosiderin deposit |
| Eczematid-like purpura of Doucas and Katepanakis | Eczematous changes with lichenification | Lower limbs | |
| Purpura annularis telangiectodes of Majocchi | Symmetric annular brown-red macules, with a clear atrophic center | Lower limbs | |
| Geugerot and Blum’s Disease | lichenified orange-red or purpuric plaques | Legs, thighs, trunk, and occasionally arms | |
| Lichen aureus | Purpuric macules with orange or golden lichenoid papules | Lower limbs | Same characteristics as lichenoid band-like dermal lymphocyte infiltrate |
| Linear capillaritis | Similar to lichen aureus with linear pattern | Lower limbs | Same as other pigmented purpuras |
This condition is also known as progressive or chronic purpuric dermatosis. Schamberg’s purpura predominates in men in their 50s and is associated with viral infections. 2 The condition typically involves the pretibial region and extends proximally, sparing the face, palmo-plantar regions. 3 Schamberg’s purpura typically presents as purpuric macules forming large plaques that acquire a brownish color described as “Cayenne pepper grains”.2,3 The lesions tend to be asymptomatic but are sometimes mildly pruritic.3
Eczematid-like Purpura of Doucas and KatepanakisEczematid-like purpura or itching purpura is described as a variant of Schamberg’s purpura 3,5 that typically affects men.5 The condition is characterized by eczematous alterations on the lower extremities associated with pruritus and secondary lichenification in patients who scratch the purpura.4,5 Similar to other types of capillaritis, the condition has a chronic course with spontaneous remission.3,5 This purpura has been associated with an allergic reaction to textiles.3,4
Purpura annularis telangiectodes of MajocchiThis condition is characterized by symmetric lesions, predominantly affecting the lower extremities with proximal extension to the buttocks.3 The lesions appear as brownish-red purpuric macules with an annular or arciform pattern2 and clear center3 that can become atrophic.7 The lesions can be confused with vasculitis, so it is important to take this into account.4 The etiology is unclear, but the condition is associated with pregnancy and venous insufficiency that worsens with textile friction.2,4 It mainly affects adolescent girls and young adult women.4,7 The lesions typically display a chronic asymptomatic course, but can be associated with pruritus.2,4
Gougerot and Blum’s diseaseLichenoid purpuric dermatosis or Gougerot and Blum’s disease affects adults between 40 and 60 years of age.2 The condition is characterized by lichenified plaques with an orangish-red or purplish color on the anterior regions of lower extremities (lower legs and thighs), trunk, and occasionally arms.2,4,8 When single, a lesion can mimic Kaposi’s sarcoma.2,4 Some cases may be associated with mycosis fungoides.2,4
Lichen aureusAlso called purpuric lichen, lichen aureus is named for to its golden color.3 Affecting young adults between 20 and 30 years of age, it is characterized by typically unilateral orange or golden purpuric macules with lichenoid papules on the lower extremities.2-4 The lesions are often chronic, lasting between 3 months and 20 years.4 The condition may be associated with drugs, trauma, or venous stasis.4 It shares histological characteristics, such as erythrocyte extravasation and perivascular infiltrate, with other types of capillaritis but characteristically presents a band-like lymphocyte infiltrate below a fine Grenz zone4 that distinguishes it from other types of capillaritis on histopathology.
Linear capillaritisLinear capillaritis, or pigmented purpuric dermatosis, looks similar to lichen aureus with a linear distribution. However, histologically, the condition does not present with lichenoid infiltrate.2 The condition is five times more common in men than women and appears on the lower extremities.9
Diagnosis, treatment, and prognosisDiagnosis is clinical. Pigmented purpuras typically exhibit an asymptomatic course with minimal pruritus, occasional pain, and normal laboratory findings.2 Some cases require a biopsy to distinguish them from other entities, such as vasculitis. On histopathology, pigmented purpuras exhibit a perivascular lymphocytic infiltrate, vascular dilation with erythrocyte extravasation, and hemosiderin deposits.
No standard therapy is available, but treatment should focus on avoiding triggers. Some treatments, such as topical steroids, griseofulvin, pentoxifylline, vitamin C, and phototherapy have reported success, with response varying between patients.5,9,10 Steroid-sparing drugs such as cyclosporine and other immunomodulators such as methotrexate have been used with good results.7 Patients can be managed conservatively with compressive stockings and lower-extremity lifting to help venous stasis.2,7,9
Pigmented purpuras have a chronic benign course. However, some cases have been associated with T-cell lymphoma.2,3,4,7 These lesions leave a post inflammatory macule that is difficult to treat.2
Cutaneous manifestations of microvascular occlusion syndromesCutaneous manifestations of microvascular occlusion syndromes are characterized by retiform purpura. Inflammatory and non-inflammatory retiform purpura should be distinguished from each other, and pertinent blood tests should be performed accordingly to narrow the diagnosis and provide appropriate treatment, given the numerous differential diagnoses. In addition, treatment varies according to the etiology. The condition is harmful if incorrectly treated as an occlusive or inflammatory syndrome.11
The differential diagnoses are numerous. This section includes some of them, but the clinician should take all other options into account. Pathophysiology is a simple method for classifying the conditions and orienting diagnosis.11
Disorders based on platelet aggregationHeparin-induced necrosisHeparin necrosis is a clinical and pathological entity that requires previous heparin exposure.11-13 The patient presents retiform non-inflammatory necrotizing purpura typically five to 11 days after exposure, but immediately if there has been previous exposure or a late hypersensitive reaction months later.11,12 Lesions typically begin as painful erythematous plaques that progress to purpura with rapid necrosis at the site or distal from heparin application with a predilection for the abdomen, thighs, and legs.11,12 A platelet plug with perivascular inflammation can be observed on histopathology.14
The condition affects 1 to 5% of adults, especially middle-aged women with a 30% incidence and mortality.11,12,15 The following pathophysiological mechanisms have been proposed: 1) heparin-induced immunity in which IgG antibodies are directed against platelet factor 4 and heparin, causing platelet plugging and consumption leading to microvascular occlusion;11-13 2) Arthus-type III hypersensitivity reaction; and 3) physical and mechanical factors, such as poor injection technique or poor adipose tissue circulation.12
It is mandatory to rule out heparin-induced thrombocytopenia or HIT syndrome to guide treatment.16 HIT characteristically presents with thrombocytopenia (30-50% of baseline value) after heparin exposure.13 Ten to 20% of patients exhibit an elevated INR value and hypofibrinogenemia consistent with disseminated intravascular coagulation (DIC) and positive serological tests, such as platelet serotonin-release assay (SRA), which is considered the “gold standard” for HIT diagnosis with 88% sensitivity and 100% specificity.13,17 Confirmation with anti-PF2/heparin antibodies by enzyme immunoassay (EIA) is also recommended.13
Treatment is based on discontinuing heparin and using an alternative anticoagulant therapy such as a thrombin inhibitor followed by vitamin K, if needed.12,15,16 Alternative heparin is reserved for cases not associated with HIT syndrome.12 Wound care, pain management, and surgical debridement and skin graft should be provided when needed.12,16
Thrombocytopenia secondary to myeloproliferative disordersMyeloproliferative disorders are characterized by abnormal proliferation of one or more cell lines on peripheral blood tests. The condition differs from acute leukemia and mainly affects young women.11 Polycythemia vera and essential thrombocytopenia are the most common forms affecting the skin.11 These conditions can present as cutaneous vascular occlusion syndromes in the form of purpura, hematomas, erythromelalgia, livedo reticularis, Raynaud’s phenomenon, leg ulcers, gangrene, and thrombophlebitis.11,18,19 The molecular pathogenesis implicated in these diseases is poorly known, but mutations in the JAK-2 protein kinase gene have been described in 90 to 95% of patients with polycythemia vera and in 50 to 70% of patients with essential thrombocytopenia.11 Platelet thrombi can be found in the dermal blood vessels of skin biopsies.20 Patients with this sign exhibit increased morbidity due to greater thrombotic or hemorrhagic potential or transformation to myelofibrosis or leukemia, compared to the general population.21 Treatment is oriented by age and cardiovascular risk. Anagrelide, low-dose aspirin, hydroxyurea, and phlebotomy have been useful in cases of polycythemia vera.21
Purpura due to cryoagglutination or cryogelling disordersCryoagglutination disorders include cryoglobulinemias, cryofibrinogenemias, and cold agglutinins.11 These syndromes are uncommon but should be taken into account for the differential diagnosis of vascular occlusion syndromes. Immunoglobulin precipitates at temperatures below 4°C and dissolves at 37°C, corresponding to cryoglobulinemias.22,23 Cryofibrinogenemias are caused by plasma fibrinogens that gel at low temperatures, and cold agglutinin antibodies promote erythrocyte agglutination at cold temperatures, causing vascular occlusion.11
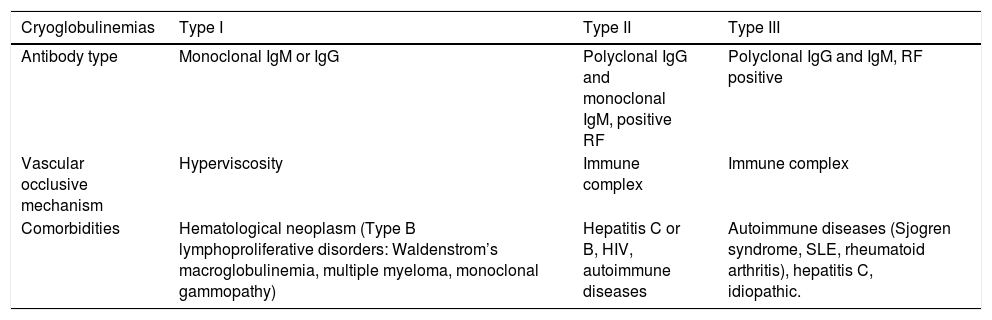
Cryoglobulinemias can be categorized into 3 types according to Brouet’s classification (chart3).22,23 Type 1 is vascular occlusive, whereas types 2 and 3 cause small vessel vasculitis due to immune complexes. These conditions affect women more frequently (at a ratio of 3:1), and the least common cryoglobulinemia is type 1.23 The etiology is based on autoimmune diseases in 10 to 15% of mixed cryoglobulinemia, hematologic neoplasms in 10 to 15% of type 1 cryoglobulinemia, and idiopathic or infectious in 25% of cases of mixed cryoglobulinemia.22 The pathogenic mechanism is based on monoclonal or polyclonal antibodies secondary to lymphoproliferative or immune stimulation due to infectious or autoimmune diseases.23,24 This section focuses on type 1 cryoglobulinemia, since the other two types cause vasculitis due to immune complexes.
Cryoglobulinemia: Classification and Characteristics
| Cryoglobulinemias | Type I | Type II | Type III |
|---|---|---|---|
| Antibody type | Monoclonal IgM or IgG | Polyclonal IgG and monoclonal IgM, positive RF | Polyclonal IgG and IgM, RF positive |
| Vascular occlusive mechanism | Hyperviscosity | Immune complex | Immune complex |
| Comorbidities | Hematological neoplasm (Type B lymphoproliferative disorders: Waldenstrom’s macroglobulinemia, multiple myeloma, monoclonal gammopathy) | Hepatitis C or B, HIV, autoimmune diseases | Autoimmune diseases (Sjogren syndrome, SLE, rheumatoid arthritis), hepatitis C, idiopathic. |
Type 1 cryoglobulinemias affect skin and other systems according to their pathophysiological mechanism of hyperviscosity or vascular occlusion due to protein precipitation. These conditions affect skin as purpura in acral areas exposed to cold temperatures, acrocyanosis, necrosis, cutaneous ulcers, Raynaud’s phenomenon, and livedo reticularis.11,22,23 Other systems are affected, and symptoms include headache, confusion, blurred vision, epistaxis, and hearing loss.22 Skin ulcers and necrosis are more common in type 1 cryoglobulinemia than in types 2 or 3.25 Diagnosis is made by clinical suspicion, presence of serum cryoglobulins on immunoelectrophoresis, immunofixation, or immunoblotting with 98, 54 and 28% sensitivity and specificity, respectively, and skin biopsy exhibiting non-inflammatory thrombosis.23,25
Prognosis is poor due to skin severity and the associated hematological disorders.22 Reported survival rates vary from 87 to 94%.26 Treatment should be individualized for patients based on their comorbidities, pathological mechanism, and disease severity.22,23 The most important measure is to avoid exposure to cold. Corticosteroids, cyclophosphamide, or biologics, and plasma exchange for severe hyperviscosity can be used in individualized cases.22,23,24
Vascular occlusion due to microorganismsThese syndromes cause vascular occlusion due to microorganisms and mainly affect immunosuppressed patients. The group includes ecthyma gangrenosum, Lucio’s leprosy, opportunistic fungal infections, and others.
Ecthyma gangrenosum is characterized by red to purple macules that form pustules or hemorrhagic ampules that evolve into necrotic ulcers within 12 to 24 hours (Figure 2).27,28 Bacterial proliferation on subcutaneous vessel adventitia forms a thrombus, causing vascular occlusion mainly in the buttocks and lower extremities.11,27,29 Absence of suppuration differentiates ecthyma gangrenosum from pyoderma gangrenosum.27 In total, 73.65% of cases of ecthyma gangrenosum are due to Pseudomonas aeruginosa, whereas 17.35% and 9% are due to other bacteria and opportunistic fungi, respectively. The former cases should be referred to as “ecthyma gangrenosum-like”.29 A compromised immune system is not an obligatory factor for the disease but has been reported in 59% of cases.29 Diagnosis is made with clinical suspicion and skin cultures. Dermal necrosis with neutrophil and lymphocyte inflammatory infiltrate and vasculitis with occlusive thrombi have been described on skin biopsy.28 Gram-negative bacteria can be found on vascular adventitia and perivascular areas. 11

Lucio’s leprosy, also referred to as necrotizing erythema of Latapi, is a type 2 reaction that corresponds to a necrotizing vasculitis with thrombosis.30 The condition is found mainly in Mexico and Central America, affecting patients with primitive lepromatous leprosy and non-nodular secondary diffuse types.30,31 It has a 5-day course initiating on the lower extremities and progressing upward until reaching the face.30,31 The lesions begin as purpuric macules surrounded by erythema that progress to bullae that necrotize.31 Skin biopsy findings depend on the stage of evolution at which the biopsy is obtained, but acid-fast bacilli with Fite-Faraco are always present.30
The treatment of vascular occlusion syndromes due to microorganisms aims to improve the patient’s immune status and treat infection. In ecthyma gangrenosum, antibiotics should be administered empirically with aggressive treatment against fungi and bacteria generally with ceftazidime, ampicillin, amoxicillin/clavulanic acid, or amphotericin B. In addition, surgical debridement is performed when indicated.29 These patients have a poor prognosis when associated with bacteremia, resulting in 20 to 50% mortality.28 For Lucio’s leprosy, thalidomide (200 to 600mg/day) should be administered until a response is observed.30 Alternatively, plasmapheresis is provided in non-responsive cases together with multibacillary treatment and high-dose prednisone (1mg/kg) with monthly tapering.31
Purpura due to embolization disordersCholesterol embolismThis condition is called “blue toe syndrome”. The disorder affects men 50 years or older, of whom 15 to 20% have a history of atherosclerotic disease, diabetes mellitus, hyperlipidemia, hypertension, and/or smoking. 11,32 Cholesterol embolisms are caused by atheromatous plaque fragmentation secondary to a force that causes plaque instability, such as cardiac catheterization, prolonged anticoagulation, antithrombotic therapy, hemorrhage, inflammation, or infection. The lesions present in hours, days, or months.11,33 In total, 20% of cases are spontaneous.32 Manifestations are noted on the skin in 35-88% of cases as well as in the kidneys and digestive tract, with a variety of cutaneous lesions, such as livedo reticularis in 40%, peripheral gangrene in 35%, cyanosis in 28%, ulceration in 17%, nodules in 10%, and purpura in 9%, along with malaise.11,32,34 Skin manifestations are bilateral and limited to the lower extremities, with normal peripheral pulses.33 Skin biopsies reveal intravascular clefts that are diagnostic in 92% of cases and correspond to cholesterol crystals that dissolve while fixing the tissue.11,32 The condition carries a poor prognosis with a high mortality rate (81%), secondary to cardiac and renal complications.34 Treatment is supportive with aspirin, statins, prostacyclin analogs, discontinuation of anticoagulation, and bypass or endarterectomy in special cases.11 The use of systemic steroids is controversial.32,33
Systemic coagulopathiesWarfarin-induced necrosisThis condition is due to abnormal γ-carboxylation of vitamin K-dependent factors, including proteins C and S, leading to a hypercoagulable state in 24 to 48 hours.11,35,36 The condition mostly occurs in women (4:1 ratio) from 60 to 70 years of age, especially in patients with congenital protein C deficiency.11,36 It has a < 0.1% incidence in treated patients, presenting in 90% of cases approximately 2 to 5 days after initiating treatment.35 Manifestations are apparent in areas of subcutaneous fat, such as the chest, abdomen, buttocks, and thighs, and are characterized by well-defined painful erythema that turns purplish and necrotic.11 Diagnosis is made by clinical suspicion and is differentiated from hematoma, disseminated intravascular coagulation, purpura fulminans, cellulitis, and calciphylaxis.37 Histopathology of the skin shows non-inflammatory thrombosis of dermal blood vessels.11 Treatment consists of the discontinuation of warfarin and administration of vitamin K. Heparin is administered in cases when anticoagulation is needed, and protein C concentrates are also provided.35-37
Purpura fulminansPurpura fulminans is a term used to describe any clinical presentation of disseminated purpura (ecchymosis, palpable, and retiform) in septic patients. Neonatal, acute, and idiopathic presentations have been described.38 Clinical lesions reflect disseminated intravascular coagulopathy due to deficiency or dysfunction of coagulation factors such as protein C, S, or antithrombin III, manifesting as distal symmetric gangrene (Figure 3).11 35,39 The lesions begin as non-blanchable, painful, distal purpuric lesions with an indurated halo evolving into bullae that turn hemorrhagic and necrotic.39 This purpura is mainly due to meningococcal infection but can be secondary to other bacterial or viral infections.11 Blood tests are consistent with disseminated intravascular coagulation.11,38 Skin biopsy reveals fibrin clots in dermal vessels with some inflammatory infiltrate.35 The case-fatality rate is high at 50%,and requires multidisciplinary treatment with blood and coagulation factor transfusions, plasma exchange with prednisone, and heparin anticoagulation.11,38
Antiphospholipid antibody syndrome
Antiphospholipid antibody syndrome (APS) causes cutaneous or systemic vessel occlusion due to anticardiolipin antibodies and circulating antiphospholipids that damage endothelial cells upon binding to exposed phospholipids and interfering with normal procoagulant protection, leading to thrombosis.11 The condition affects the skin in 70% of cases, presenting as a variety of skin lesions, including livedo reticularis and gangrene. A rare catastrophic variant is noted in 1% of cases with a 50% mortality rate.11,40 The condition mainly affects women between 15 and 50 years of age.11 Testing for anticardiolipin antibodies can be ordered upon suspicion, and anti-ß2 glycoproteins are more specific. In addition, the lupus anticoagulant and Venereal Disease Research Laboratory tests (VDRL) show positive results.11,40 Histopathology of skin lesions reveals non-inflammatory thrombosis of dermal vessels.41 Treatment is based on clinical history and patient risk. Anticoagulation and antiplatelet drugs are administered in low-risk patients, and high-dose systemic steroids with anticoagulation, intravenous gamma globulin, and plasma are administered in high-risk patients.40,42
Others: CalciphylaxisCalciphylaxis, also known as calcifying panniculitis or calcific uremic arteriolopathy, is a lethal disease that affects chronic renal patients on hemodialysis and hyperparathyroidism patients, with 80% case-fatality rate.11, 43-45 Manifestations include calcification of the arterial midlayer and subintimal fibrosis followed by thrombotic occlusion.43 The condition affects areas such as abdomen, buttocks, and thighs, with varied clinical manifestations including livedo reticularis, plaques, or painful violaceous subcutaneous nodules with necrotic ulcers and eschar with superinfection in some cases (Figure 4).44 Skin biopsy reveals Von Kossa-positive calcium deposits in the arterial midlayer, intimal fibrosis, and intraluminal thrombus along with erythrocyte extravasation.45,46 Treatment should be aggressive and include metabolic monitoring of calcium, phosphate, and parathyroid hormone levels. Treatment is based on phosphate binders, a phosphorus-free diet, discontinuation of vitamin D, and antibiotics in case of superinfection.43 Other treatments include intravenous sodium thiosulfate as a calcium binder, bisphosphonates, cinacalcet, low- calcium dialysis, hyperbaric oxygen therapy, and parathyroidectomy.11,43,44 Steroids are spared due to superinfection risk but can be used in early-stage cases without necrosis.43,44 The diagnosis carries a poor prognosis.
CONCLUSION
Purpura is one of the most frequent conditions seen in dermatology practice and covers a wide range of differential diagnoses. It is important to consider all differential diagnoses and to know their basic pathophysiology because treatment varies greatly according to etiology, despite the fact that the biopsy and clinical manifestations may seem very similar. Vasculitis is also within the spectrum of differentials. However, this very important and extensive topic is not included in this review, but we should not forget to take this condition into account when approaching a patient with purpura.
Financial support: None.
Conflict of interest: None.
