









Bullous pemphigoid is the most frequent autoimmune bullous disease and mainly affects elderly individuals. Increase in incidence rates in the past decades has been attributed to population aging, drug-induced cases and improvement in the diagnosis of the nonbullous presentations of the disease. A dysregulated T cell immune response and synthesis of IgG and IgE autoantibodies against hemidesmosomal proteins (BP180 and BP230) lead to neutrophil chemotaxis and degradation of the basement membrane zone. Bullous pemphigoid classically manifests with tense blisters over urticarial plaques on the trunk and extremities accompanied by intense pruritus. Mucosal involvement is rarely reported. Diagnosis relies on (1) the histopathological evaluation demonstrating eosinophilic spongiosis or a subepidermal detachment with eosinophils; (2) the detection of IgG and/or C3 deposition at the basement membrane zone using direct or indirect immunofluorescence assays; and (3) quantification of circulating autoantibodies against BP180 and/or BP230 using ELISA. Bullous pemphigoid is often associated with multiple comorbidities in elderly individuals, especially neurological disorders and increased thrombotic risk, reaching a 1-year mortality rate of 23%. Treatment has to be tailored according to the patient’s clinical conditions and disease severity. High potency topical steroids and systemic steroids are the current mainstay of therapy. Recent randomized controlled studies have demonstrated the benefit and safety of adjuvant treatment with doxycycline, dapsone and immunosuppressants aiming a reduction in the cumulative steroid dose and mortality.
The term pemphigoid was first introduced by Lever in 1953 to describe a disease characterized by bullous formation due to subepidermal detachment in order to distinguish it from pemphigus, an intraepidermal blistering disorder induced by acantholysis.
Subsequently, Jordan and Beutner demonstrated the presence of autoantibodies in patients with bullous pemphigoid (BP) using direct and indirect immunofluorescence techniques.1
EpidemiologyBP is the most frequent autoimmune blistering disorder.2 It affects mainly elderly individuals, during the 8th decade of life, without gender predilection, with rare case reports of BP in children and adolescents.3,4
The incidence of BP has increased over the past decades as a result of population aging with multiple comorbidities and exposure to drugs that may potentially trigger the disease, as well as improvement in the clinical diagnosis of non-bullous presentations and in the accuracy of laboratory techniques to demonstrate the presence of autoantibodies against hemidesmosomal proteins.3
Epidemiological studies of BP in Europe demonstrate the incidence ranges from 2.5 to 42.8 cases/million/year, whereas in Asia the annual incidence is estimated in 2.6 to 7.5 cases/million.5-9 Statistical differences may be related to the type of study performed (retrospective vs. prospective), duration and collected data (national databases, chart review). The frequency of BP also increases in patients older than 80 years, with nearly 150-330 new cases/million/year at this age range.3
PathogenesisBP is characterized by autoantibodies that recognize self-antigens at the basement membrane zone (BMZ), known as BP180 (180kDa) or BPAG2, and BP230 (230kDa) or BPAG1. Both antigens are key components of the hemidesmosome, which is responsible for the adhesion between the epidermis and dermis.3
BP230 is an intracellular component of the hemidesmosome that belongs to the plakin family of proteins. IgG autoantibodies react against globular C-terminal domains of BP230.3
BP180 is a transmembrane glycoprotein of nearly 1,500 amino acids with an extracellular domain – NC16A – the main antigenic epitope in BP. In addition to NC16A, patients with BP also develop IgG autoantibodies directed against other epitopes; reactivity against C-terminal and intracellular epitopes are related to mucosal involvement during the early stages of the disease.3
Once anti-NC16A autoantibodies bind to BP180, several pathways are activated, including complement activation and deposition, neutrophilic chemotaxis with release of proteases and elastases that promote the disruption of the BMZ leading to blister formation.2
IgG1 and IgG3 are the predominant circulating anti-NC16A IgG subclasses followed by IgG4. Studies performed in skin sections of humanized NC16A mice demonstrated that both IgG1 and IgG3 are able to bind to the BMZ and fix complement. On the other hand, concomitant incubation with IgG4 promotes its deposition at the BMZ and prevents IgG1 binding thus inhibiting complement fixation, neutrophil chemotaxis and blister formation. Similar results were obtained with the injection of anti-NC16A IgG1, IgG3 and IgG4 into humanized NC16A mice.10
Disease activity correlates with circulating levels of anti--BP180 IgG and potentially to serum levels of anti-BP180 IgE.11 Anti-BP180 IgA and IgE have also been described. Xenograft mice models of human skin injected with hybridomas containing IgE against LABD97 or IgE isolated from patients with BP develop intense dermal infiltration of eosinophils, neutrophils and mast cells with disruption of the BMZ and subepidermal detachment.12 Eosinophilic infiltration is a main histopathologic finding in BP. Lin et al. demonstrated in a humanized IgE receptor mouse model that eosinophils are necessary for IgE-mediated blister formation in BP, providing the cellular link between IgE autoantibodies and skin lesions in the murine model.13
Messingham et al. proposed two IgE-mediated mechanisms of blister formation: IgE may interact with the FcεRI receptors on mast cells and promote their cross-linking through binding of the NC16A domain of BP180, followed by the degranulation of histamine and cytokines and chemotaxis of eosinophils and neutrophils; in addition, IgE may also bind directly to the NC16A domain of BP180 expressed on keratinocytes; the internalization of this immune complex leads to the release of IL-6 and IL-8, which recruit additional immune cells.14 There is no report of consistent association between serum levels of anti-BP180 IgE and a specific clinical manifestation of BP such as the presence of urticarial lesions.11
Neurologic disorders and BpBoth BP and neurological diseases affect elderly individuals with multiple comorbidities under the use of several medications,
and epidemiological studies provided evidence that their coexistence is not coincidental. A systematic review with meta-analysis evaluated 14 studies with 23,369 BP patients and 128,697 controls. This review indicates that BP patients are 5 times more prone to develop any neurologic disorder, mainly multiple sclerosis, dementia, Parkinson’s disease, epilepsy and stroke, which usually precedes the onset of BP by 5.5 years.15 Multiple sclerosis has the highest association, with a 5-12 time risk of development of BP.15,16
The pathogenic processes that link the development of BP and neurologic disorders are not fully understood. Experimental studies demonstrated that bullous pemphigoid antigen (BPAG1 and BPAG2) are expressed in the skin and central nervous system.17 It is believed that an insult to the central nervous system may trigger the exposure of antigens such as neuronal BP180 followed by the synthesis of anti-BP180 IgG. Levels of circulating anti-BP180 autoantibodies even correlate with the severity of dementia in patients with Alzheimer’s disease.18 Due to an epitope-spreading phenomenon, these neuronal autoantibodies may also cross-react with cutaneous BP180 and precipitate the onset of BP.19,20
Malignancies in BpThe association of malignancies and BP have conflicting data. A Japanese study with 1,113 BP patients showed 5.8% of malignancies (gastric, colorectal, lung prostate and uterine cancers and lymphomas), higher than the expected for age-matched controls.21 Another Japanese review of 115 BP patients revealed 10.4% of internal malignancies (gastric, colorectal, renal, bladder, prostate, laryngeal, lung and breast cancers), higher than the expected incidence for the general Japanese population.22 A Singapore study with 359 BP patients showed no increased incidence of malignancies.23 A German study with 8.3 million subjects showed 6.7% of hematologic malignancies (Hodgkin lymphoma, non-follicular lymphoma, mature T/NK-cell lymphoma, non-Hodgkin lymphoma, myeloid leukemia, and other leukemias) in 1,743 BP patients with no association with non-hematologic malignancies.24
A systematic review and meta-analysis of BP and malignancies including 8 studies (1 retrospective cohort, 2 case-controls and 5 cross-sectional studies) found no association of BP with overall malignancy; however, a possible association with hematologic malignancies was observed.25 An English study in a cohort of 2,873,720 individuals with malignant neoplasms showed no overall greater risk of concurrent or subsequent BP than individuals with no record of cancer. However, in sub-cohorts of individuals with either kidney/laryngeal cancer or lymphoid leukemia there was elevated risk for BP.26
Thrombotic risk and BpBP is an autoimmune condition that promotes a dysregulated immune response mediated by Th1 and Th2 cells, with increased synthesis of IL-1β, TNF-α, IL-5, IL-6, IL-8, IL-10 and IL-15.3 Such pro-inflammatory cytokines induce a systemic response with upregulation of vascular endothelial growth factor and E-selectin resulting in endothelial cell activation.27 Additionally, BP patients with active lesions exhibit increased circulating levels of D-dimer and prothrombin and overexpression of tissue factor in lesional skin that return to normal levels upon disease control.28 Tissue factor is protein expressed in eosinophils that binds the factor VIIa and acts as a key activator of the extrinsic coagulation pathway.29 This prothrombotic state during BP activity translates into an increased risk of thromboembolic events including pulmonary embolism (adjusted OR 3.12) and stroke (adjusted OR 2.37) in comparison to age-matched controls.30,31
Clinical Aspects and ClassificationThere is currently no standardized classification of BP. However, it is possible to recognize distinct variants of the disease according to the age of onset, clinical presentation and triggering factors.
Classic BpClassic BP affects elderly individuals, usually above 70 years old and presents with itchy, tense blisters over normal skin or over erythematous and edematous background on the trunk and extremities. These lesions affect mainly the axillary folds, lower abdomen, inguinal areas and inner parts of the thighs. They may be localized or widespread. Mucosal involvement is not often detected, and is reported in 10-30% of the cases with oral, esophageal and genital lesions (Figure 1).
 and hyaline (B) content on the trunk and limbs with erythematous and edematous background; hyaline blisters without inflammatory signs (C); excoriated papules and blisters with crusts in the axillary region (D); brownish and erythematous plaques with overlying purulent and hyaline blisters (E). Mucosal involvement - blisters and erosions on the palate (F); blister in the esophagus (G)")
Classic bullous pemphigoid – Tense blisters with hemorrhagic (A) and hyaline (B) content on the trunk and limbs with erythematous and edematous background; hyaline blisters without inflammatory signs (C); excoriated papules and blisters with crusts in the axillary region (D); brownish and erythematous plaques with overlying purulent and hyaline blisters (E). Mucosal involvement - blisters and erosions on the palate (F); blister in the esophagus (G)
One of the clinical presentations of BP is lichen planus pemphigoid. Patients with this condition may present pruritic violaceus plaques and papules, resembling lesions of lichen planus, usually on the extremities. Later on, blisters and vesicles appear over lichenoid lesions and over normal skin. Buccal mucosa may present whitish, lace-like, reticulated lesions.32
Nonbullous BpReports of BP without blisters have raised the question whether nonbullous BP should be considered as a variant of the disease instead of a prodromic phase. According to a systematic review published in 2017, only 9.8% of the patients who present with nonbullous BP at the onset of the disease develop blisters during their follow-up, and rarely display mucosal lesions. The most frequent clinical findings include pruritus (100%) with erythematous urticarial lesions (52.3%), excoriations (22.7%), or papules and nodules (20.5%)33 (Figure 2).
. Erythema multiforme-like: targetoid plaques on the thighs (B). Prurigo-like: lichenified papules on the dorsum of the hand (C). Excoriations on the frontal region (D). Eczematous: erythematous-brownish confluent plaques on the trunk and upper limbs (E). Toxic epidermal necrolysis-like: erosions on the dorsum (F)")
Nonbullous pemphigoid – Urticariform: confluent erythematous and edematous papules on the trunk (A). Erythema multiforme-like: targetoid plaques on the thighs (B). Prurigo-like: lichenified papules on the dorsum of the hand (C). Excoriations on the frontal region (D). Eczematous: erythematous-brownish confluent plaques on the trunk and upper limbs (E). Toxic epidermal necrolysis-like: erosions on the dorsum (F)
Another rare manifestation of BP may be exfoliative erythroderma. Generalized erythema and desquamation even in the absence of blisters are seldom reported. Clinical suspicion is challenging in this situation as other disorders that more frequently present as erythroderma such as pemphigus foliaceus, psoriasis, eczema and drug reactions are common misdiagnoses (Figure 3).34
Infantile and childhood Bp
BP may occasionally affect infants and children, with nearly 100 reported cases. Though the autoantibodies in younger patients with BP recognize the same epitopes within the non-collagenous 16A domain of BP180 as in classic BP, differences in the clinical course and response to therapy have been described.35
Two peaks of incidence, one with an average age of 4 months in infantile BP and another with an age of 8 years in childhood BP were characterized. Infantile BP significantly affects the face (62%), palms and soles (79%), with an increased frequency generalized lesions that prompt use of systemic corticosteroid with good response36-39 (Figure 4).
. Disidrosiform vesicles on the hands (B)")
Vaccination has been considered as a potential trigger of BP is this age group since some reported cases occurred hours or days following vaccine administration and even experienced spontaneous remission.39 Nevertheless, a precise and final correlation between vaccination and BP development has not been demonstrated.40 On the other hand, childhood BP presents localized lesions with genital involvement observed in 17% of the cases with remission after topical steroid treatment and/or sulfonamides (sulfapyridine or dapsone).36
Triggering Diseases and Factors in BpPsoriasis/Phototherapy-induced BpIn 1976 a series of 4 cases of psoriatic patients with confirmed diagnosis of BP was published.41 In most case reports psoriasis preceded the onset of BP in about 20 years.42 A combination of chronic damage to the BMZ mediated by neutrophilic elastases and matrix metalloproteinases, the recruitment of activated lymphocytes, and the occurrence of numerous antigen presenting cells may lead to self-antigen exposure, and prompt autoimmune mechanisms that induce autoantibody synthesis against hemidesmosomal proteins, such as BP180 and BP230.42-44
Psoriasis treatment with phototherapy may also trigger the exposure of BP antigens, and modification T-helper and T-suppressor lymphocytic responses induced by ultraviolet radiation, contributing to the occurrence of epitope spreading and autoantibody production (Figure 5).42,45,46
Lichen planus as a triggering disease for Bp and lower (B) limbs; psoriatic plaque on the thigh (C)")
Patients with lichen planus (LP), without IgG anti-BP 180 NC16A may later develop clinical and laboratory findings of BP, suggesting that damage to the basal cells may expose sequestered antigens, which may lead to the development of BP.47 It is considered an epitope spreading phenomenon triggered by chronic inflammation adjacent to the dermal epidermal junction leading to the exposure of new epitopes.48 BP may occur with bullae arising over lichenoid lesions or normal-appearing skin mainly involving the extremities, typical findings of lichen planus pemphigoid (LPP) (Figure 6). One of the remarkable differences between bullous lichen planus (BLP – variant of LP) and LPP (variant of BP) is the presence of bullae only over lichenoid lesions in BLP and bullae arising over lichenoid lesions and normal skin in LPP.
Drug-induced Bp and lower (B) limbs, and feet (C). Lichen planus pemphigoid – Confluent macules and papules on the trunk (D) and lower limbs (E) with tense hyaline blisters on the feet (F)")
BP may also be drug-induced, as above mentioned, and present a better prognosis with a good response to therapy after withdrawal of the suspected medication and fewer or no recurrences.49
Although no specific gene mutation has been identified, it is hypothesized that genetic predisposition plays a role in drug-induced BP. Genetically susceptible individuals exposed to certain medications may experience a deregulation of the immune system with inactivation of regulatory T-cells and stimulation of B-cell clones that recognize self-antigens and induce autoantibody production.49 Several drugs may induce BP, such as inhibitors of dipeptidyl peptidase-IV (DPP-IV), diuretics, antipsychotics, checkpoint inhibitors and others, as shown in chart 1.50-53
Drugs reported to induce bullous pemphigoid
| Drug class | Examples | Study design | OR |
|---|---|---|---|
| Dipeptidyl peptidase-IV (DPP-IV) inhibitors | Vildagliptin | Systematic review and meta-analysis including 3563 BP patients using DPP-IV inhibitors50 | Pooled 10.16 |
| Linagliptin | Pooled 6.13 | ||
| Diuretics | Furosemide | British case-control study including 86 patients with BP and 134 age and sex-matched controls51 | Adjusted 3.8 |
| Spironolactone | French case-control study with 201 BP patients and 345 controls matched according to age, sex, place of residence and hospital52 | Adjusted 2.3 | |
| Antipsychotics | Phenotiazine aliphatic chain | Adjusted 3.7 | |
| Checkpoint inhibitors Anti-PD-1/PD-L1 | Pembrolizumab, Nivolumab, Durvalumab | Review study including 21 case reports of BP induced by checkpoint inhibitors53 | Not applicable |
No specific biomarker of drug-induced BP has been recognized, and the clinical presentation and immunopathological findings may be indistinguishable from the classic form. Onset of the disease at a younger age and within an average of 3 months after the introduction of a new drug, with rapid disease control with fewer or no recurrences once the culprit medication is withdrawn may suggest its role as a triggering factor.49
Dipeptidyl-peptidase IV inhibitorsDPP-IV inhibitors are oral hypoglycemic agents approved by the Food and Drug Administration in 2006. Keratinocytes also express DPP-IV, which modulates cytokine synthesis and glucagon pathways observed in skin structures. As a result, exposure of new epitopes of BMZ combined with an altered immune response to self-antigens may prompt BP development.54
A systematic review and meta-analysis including 3,563 patients demonstrated a 3.6 fold risk of BP in patients receiving DPP-IV inhibitors, with a higher association with vildagliptin (pooled OR=10.16; CI=6.74-15.33) and linagliptin (pooled OR=6.13; CI=2.51-15.0).50 The presence of HLA-DQB1*03-01 poses a risk factor for BP induced by DPP-IV inhibitors with 86% sensitivity and 69% specificity. Patients with this allele often present bullae without prominent inflammatory signs.55 In the majority of BP patients, withdrawal of the DPP-IV inhibitors and treatment with high-potency topical steroids lead to disease remission.54,56
Checkpoint inhibitorsSince the advent of checkpoint inhibitors that target the programmed cell death protein-1 (PD-1) and the programmed death ligand-1 (PD-L1), a growing number of patients with metastatic malignancies has been receiving immunotherapy as an adjuvant treatment. Monoclonal antibodies that target PD-1/PDL-1 pathways may induce immune-mediated adverse events possibly related to a reduction in regulatory T cells leading to increased T-cell activation, B-cell proliferation, and synthesis of autoantibodies.57
Cutaneous adverse events are reported in nearly 50% of the patients receiving immunotherapy, and range from pruritus and exanthematous reactions to autoimmune blistering disorders.58 A review published in 2018 included 21 case reports of BP induced by checkpoint inhibitors with a male predominance (15/21) and mean age at diagnosis of 71 years.53 The most frequent PD-1 and PD-L1 inhibitors associated with BP are pembrolizumab, nivolumab and durvalumab.53,57,59,60
Pruritus may be a prodromal sign of BP development and occur between 19-21 weeks after the introduction of PD-1/PD-L1 inhibitor, while bullae may only appear within 20-39 weeks of therapy. Most patients were treated with topical or systemic steroids; however, two patients received biologics (rituximab and omalizumab) as steroid-sparing agents.53 Given the small number of current reports, it is not possible to determine a correlation between immunotherapy dose or cancer progression and BP onset and prognosis at this point.
Clinical DiagnosisBP should be considered as a differential diagnosis of pruritus in elderly patients, regardless of the presence of blisters.33 Once nonbullous BP and classic BP have similar age of onset, distribution of lesions and immunological characteristics, BP should be considered as a differential diagnosis in patients over 70 years of age with chronic pruritus without an underlying systemic cause. The diagnosis can be confirmed by direct or indirect immunofluorescence studies with a specificity of nearly 100%.33
Clinical evaluation of a patient with suspected BP involves a thorough dermatological examination of the skin and mucosa. There are two disease scores that are currently available for measuring BP activity, BP Disease Area Index (BPDAI) and Autoimmune Bullous Skin Disorder Intensity Score (ABSIS).61,62 Comparison of these two BP activity scores showed that BPDAI demonstrated excellent reliability, validity and responsiveness.63
Referral to other specialties such as Ophthalmology, Ear Nose and Throat, Gynecology and Gastroenterology is of great relevance to the evaluation of potential mucosal involvement.
Introduction of new drugs or presence of other triggering factors such as radiotherapy and phototherapy are additional clues for the diagnosis.49Assessment of possibly related diseases including neurologic disorders and psoriasis is also recommended.
Laboratory DiagnosisDiagnosis relies on a careful correlation of clinical features suggestive of BP with immunopathological findings. Histopathology of non-bullous lesions usually demonstrates the presence of eosinophilic spongiosis with a mixed dermal inflammatory infiltrate; bullous lesions are usually characterized by subepidermal detachment with eosinophils, neutrophils and fibrin in the blister content, and dermal inflammatory infiltrate (Figures 7A and 7B).33
; B. Subepidermal detachment filled with eosinophils and fibrin (Hematoxycilin & eosin, x20). Immunofluorescence characteristics in bullous pemphigoid – C. Linear C3 deposition at the BMZ; D. IgG bound to the epidermal side of the detachment using the salt-split skin technique")
Histopathological features of bullous pemphigoid – A. Eosinophilic spongiosis with eosinophilic dermal infiltrate (Hematoxycilin & eosin, x20); B. Subepidermal detachment filled with eosinophils and fibrin (Hematoxycilin & eosin, x20). Immunofluorescence characteristics in bullous pemphigoid – C. Linear C3 deposition at the BMZ; D. IgG bound to the epidermal side of the detachment using the salt-split skin technique
In situ and circulating autoantibodies against hemidesmosomal proteins can be detected using direct and indirect immunofluorescence (IF), respectively. Direct immunofluorescence (DIF) is performed in a sample obtained from perilesional skin; linear deposits of IgG and/or C3 along the BMZ are consistent with the diagnosis of BP (Figure 7C). Indirect immunofluorescence (IIF) on monkey esophagus exhibit linear IgG deposition at the dermal epidermal junction, and using salt-split human skin as substrate the deposition occurs on the epidermal side of the split (Figure 7D).
Serum anti-BP180 and anti-BP230 autoantibodies may also be quantified using ELISA, immunoprecipitation and immunobloting. A retrospective study including 313 patients with BP and 488 controls compared the sensitivity and specificity of DIF, IIF and ELI-SA for the diagnosis of BP. Among all methods, DIF displayed the highest sensitivity similarly to all combined serologic tests (90.8% vs. 88.8%; p=0.56).64
Nemeth et al. established clinical and immunopathological criteria for the diagnosis of childhood BP including age up to 18 years: presence of tense blisters with or without underlying erythema or mucosal involvement, histopathological confirmation of subepidermal detachment with eosinophils, and demonstration of IgG and/or C3 deposits at the BMZ using direct or indirect immunofluorescence.65
Differential DiagnosisAs BP may have a polymorphic presentation with non-bullous manifestations and blisters, a wide range of differential diagnosis should be considered: pemphigus foliaceus, pemphigus herpetiformis, linear IgA bullous dermatosis, epidermolysis bullosa acquisita, bullous lupus erythematosus, eczema, urticaria, prurigo, impetigo, erythema multiforme, Sweet syndrome, toxic epidermal necrolysis and autotoxic pruritus.
Evolution And ComplicationsBP is a chronic relapsing disease that affects elderly individuals often with multiple comorbidities and functional impairment.66 A correlation between disease activity and serum levels of anti-BP180 IgG3 and IgE was demonstrated and these may be useful biomarkers to guide steroid tapering and to determine the set of patients that can benefit from the treatment with omalizumab.67
Complications related to the disease, such as secondary infection leading to hospitalization and sepsis, or to adverse effects of the medications required for disease control translates into a 3.6-fold higher mortality than age-matched population.68 The most frequent primary discharge diagnoses for BP inpatients in the US were septicaemia, pneumonia and urinary tract infection.69
A multicentric retrospective study of 801 patients with autoimmune blistering diseases found one patient with Pneumocystis pneumonia (PCP), resulting in an incidence rate of 0.1%, bellow the recommended threshold of 3.5% to justify prophylaxis for PCP. This study has several limitations, due to its retrospective nature, prior use of antibiotics and use of different medications and doses. The above-mentioned patient with PCP did not present lymphopenia, neutropenia or other pulmonary abnormalities. However, it should be emphasized that patients under immunosupressive therapy should be monitored, in order to identify lymphopenia, prompting either a switch in their treatment or temporary PCP prophylaxis.70
PrognosisRisk factors related to increased mortality in BP include older age, neurological disorder (dementia, including Alzheimer’s disease and other types of dementia, and stroke), and increased serum levels of anti-BP180 NC16A IgG.71,72
A recent systematic review and meta-analysis included 25 studies and demonstrated a 1-year combined mortality rate of 23.5%. Higher mortality rates were obtained in Europe (26.7%) followed by Asia (20.5%) and US (15.1%), possibly related to an increased number of comorbidities and older age in European patients in comparison to Americans and Asians, respectively.73-75.
Evidence from two randomized controlled trials evaluating the use of azathioprine or dapsone and doxycycline as steroid-sparing adjuvant therapy in BP demonstrated a reduction in the 1-year mortality rate to 8% and 13.4%, respectively. 76,77
TreatmentTreatment of BP aims to arrest the development of new lesions and enable cutaneous healing and control of pruritus. As BP mainly affects elderly individuals, the choice of therapy has to be tailored according to the patient’s comorbidities and ability of self-care in order to avoid potential complications and increased morbidity and mortality.
A Cochrane systematic review published in 2010 analyzed 10 randomized clinical trials of bullous pemphigoid treatment including 1049 patients.78 Two of them evaluated the use of 0.05% clobetasol propionate cream: one in comparison to 0.5-1.0 mg/kg/ day of prednisone while the other included two different doses of the topical steroid (40g/day vs. 10-30g/day).79,80 Clobetasol cream provided an effective control of moderate disease, regardless of the daily dose with a decrease in the occurrence of severe adverse effects and mortality in relation to oral steroid.
Nevertheless, some limitations related to the use of topical clobetasol include the difficulty to perform self-applications of the cream to the entire body, which may also have systemic side effects and induce worsening of the cutaneous atrophy commonly observed in the skin of elderly patients.81 For these reasons, systemic steroids remain as first line treatment in some guidelines of BP treatment especially in severe cases.82-84 Though Morel et al. recommended to avoid the use of higher doses of prednisolone or prednisone than 0.75mg/kg/day because of the potential increase in the risk of adverse events and mortality, there are some limitations concerning this finding.85 Despite the fact that the results demonstrated a significantly lower mortality after a short follow-up of 51 days (3/22 deaths with 1.25mg/kg/day vs. 2/24 with 0.75mg/kg/day), an early evaluation of clinical improvement after 21 days favored the use of higher doses of corticosteroid (51% improvement with 0.75mg/kg/day vs. 64% improvement with 1.25mg/kg/day). Kirstchig also criticized the lack of blinding of the investigators and incomplete publication of outcome data. Moreover, there is no meta-analysis evaluating the systemic corticosteroid dosage due to the lack of studies with similar and well established methodology.78
Treatment with systemic steroid and azathioprine, mycophenolate mofetil, plasma exchange or the combination of tetracycline and nicotinamide showed no improvement in disease control; therefore, new studies have further evaluated the role of adjuvant therapies to reduce the cumulative dose of systemic steroid or even replace the steroid treatment as the standard of care.78
A randomized controlled study published in 2017 compared the efficacy and safety of azathioprine 1.5-2.5mg/kg/day (n=27) or dapsone 1.5mg/kg/day (n=27) as adjuvant drugs in combination with methylprednisolone 0.5mg/kg/day for the treatment of adult patients with BP.76 Though the initial sample size was not achieved and the low number of patients included in the study prevented the evaluation of the primary outcome, dapsone enabled a significantly lower steroid cumulative dose with a similar number of adverse events and complete remission rate to azathioprine. A lower 1-year mortality rate of 8% occurred in contrast to 10-40% rates obtained with exclusive steroid treatment, thus reinforcing the beneficial effect of adjuvant drugs with steroid-sparing effect.
Azathioprine is a derivative of 6-mercaptopurine that interacts with cellular DNA promoting apoptosis, B-cell proliferation arrest and reduction of antibody synthesis.83 The 6-mercaptopurine can be metabolized after enzyme steps to its active forms (6-thioguanine nucleotides), and inactivated by xanthine oxidase or thiopurine methyltransferase (TPTM). The recommended dose of azathioprine is 0.5-3mg/kg/day, when patients present normal TPTM test; dose of azathioprine is calculated according to TPTM activity.86 Even with normal TPTM test, patients under azathioprine should regularly perform complete blood cell count, liver enzymes, and evaluate potential infections and neoplasias. BP patients are usually treated with azathioprine 1.0-2.0mg/kg/day.
Dapsone reduces the release of IL-8 by cultured human keratinocytes after exposure to anti-BP180 IgG in a dose-dependent manner.76 This results in the inhibition of neutrophilic chemotaxis and activation, reducing the release of leukotrienes and prostaglandins.87
Doxycycline has been used as an adjuvant therapy in BP due to its anti-inflammatory properties without an overt immunosuppression. Inhibition of matrix metalloproteinases and neutrophilic activation caused by immune complex formation between IgG autoantibodies and BP antigens prevents the disruption of the dermal epidermal junction and may contribute to disease control.88 Williams et al. demonstrated, in a randomized, controlled trial, a non-inferiority of doxycycline 200mg/day to achieve disease control after 6 weeks of treatment in comparison to prednisolone 0.5mg/kg/day, with similar improvement of quality of life. Patients receiving doxycycline experienced significantly less adverse effects with a decreased risk of death.77 Limitations of this study include: a non-inferiority outcome (considering that doxycycline can be 25-37% inferior as treatment in comparison to prednisolone); the short period of 6 weeks of treatment (subsequent follow-up of up to 52 weeks showed higher efficacy of prednisolone); concomitant use of an unspecified amount of topical clobetasol in the initial 3 weeks of treatment in both groups, which could have had a systemic effect and accounted for the improvement observed in the doxycycline group, as well as the increased mortality in the prednisolone arm.89
Data obtained from animal models of BP demonstrated that intravenous immunoglobulin (IVIg) saturates the IgG receptor FcRn promoting anti-BP180 IgG clearance.90 Anti-idiotypic antibodies present in the IVIg also neutralize anti-BP180 antibodies and prevent immune complex formation and dermal epidermal detachment. Furthermore, release of pro-inflammatory cytokines such as IL-1 and IL-6 is reduced whereas the production anti-inflammatory IL-10 increases, contributing to disease control.91 A randomized controlled trial analyzed IVIg as an adjuvant therapy in patients with active BP without improvement after treatment with prednisolone 0.4mg/kg/day. Participants (n=56) were randomized to receive either IVIg 400mg/kg/day (n=27) or placebo (n=29) for 5 consecutive days. IVIg significantly improved disease activity until day 29 (p<0.05) of treatment with a lower daily dose of prednisolone than the placebo group (p=0.042), and a reduction in the levels of anti-BP180 antibody (p<0.05).92
Biologics with a more specific target implicated in the pathogenesis of BP have also emerged as alternative treatment. Rituximab is a medication from chimeric IgG monoclonal antibodies against CD20 – a cell-surface antigen expressed in B-lymphocytes – that induces apoptosis of self-reactive cells without affecting memory cells.93 Inhibition of the synthesis of all IgG subclasses including autoantibodies against BP180 enables disease control and tapering of systemic steroids.94
A limited number of patients received rituximab for the treatment of BP with different doses and intervals of infusions, and a wide range of concomitant steroid and/or immunosuppressive drugs, which preclude an evidence-based recommendation of use. Current data demonstrates clinical remission occurs in up to 70% of patients and is not sustained in 20% of them.92 Adverse events are also common (11-20%) including infections (11-30%) and increased mortality (11-15%).95
A protocol of treatment combining 12 infusions of rituximab over a 6-month interval along with intravenous immunoglobulin infusions was proposed by Ahmed et al., in an attempt to reduce disease recurrences and prevent infections complications. Despite the multiple infusions of rituximab and intravenous immunoglobulin, 2 out of 10 patients experienced 1 recurrence each after the end of the protocol and required additional rituximab infusions. All patients remained disease-free thereafter with negative direct immunofluorescence and salt-split skin indirect immunofluorescence after 5 years.95
Omalizumab is a medication produced from humanized monoclonal anti-IgE antibodies that binds to circulating IgE and reduces the expression of its receptor on immune cells.12 Recent studies demonstrated the role of IgE in the pathogenesis of BP. Increased IgE levels are detected in 77% of patients with and 41% have IgE deposits at the BMZ.96,97 To the best of our knowledge, 14 case reports of the use of omalizumab in patients with severe or recalcitrant bullous pemphigoid and increased serum levels of IgE and/or eosinophilia have been published. Different dose regimens and the concomitant use of systemic steroid and immunosuppressants are important limitations to establish recommendations about the use of omalizumab based on the current reports.
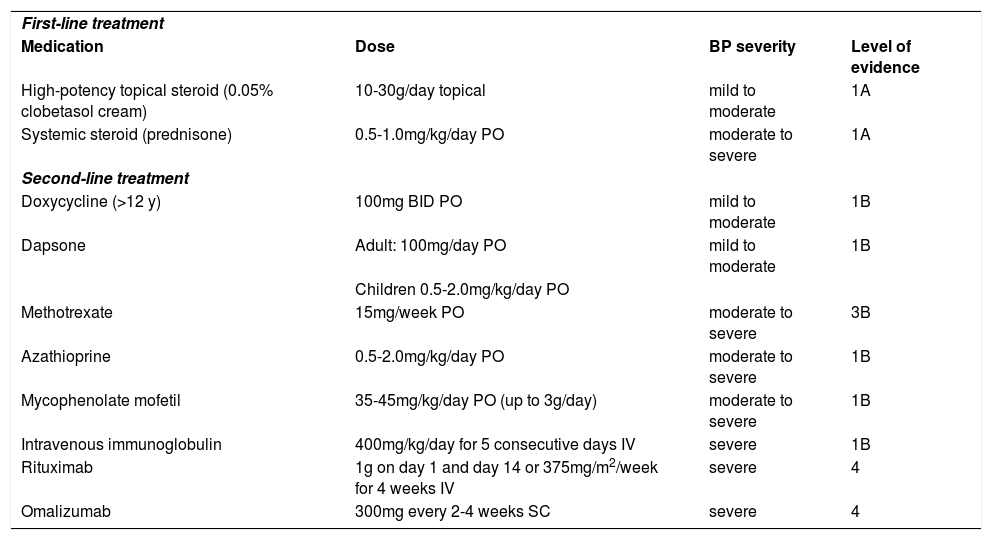
Table 1 summarizes the current evidence of BP treatment (levels of evidence according to the Centre for Evidence-Based Medicine, Oxford) (www.cebm.net).78,83,84,98
Evidence-based treatment for bullous pemphigoid according to disease severity
| First-line treatment | |||
| Medication | Dose | BP severity | Level of evidence |
| High-potency topical steroid (0.05% clobetasol cream) | 10-30g/day topical | mild to moderate | 1A |
| Systemic steroid (prednisone) | 0.5-1.0mg/kg/day PO | moderate to severe | 1A |
| Second-line treatment | |||
| Doxycycline (>12 y) | 100mg BID PO | mild to moderate | 1B |
| Dapsone | Adult: 100mg/day PO | mild to moderate | 1B |
| Children 0.5-2.0mg/kg/day PO | |||
| Methotrexate | 15mg/week PO | moderate to severe | 3B |
| Azathioprine | 0.5-2.0mg/kg/day PO | moderate to severe | 1B |
| Mycophenolate mofetil | 35-45mg/kg/day PO (up to 3g/day) | moderate to severe | 1B |
| Intravenous immunoglobulin | 400mg/kg/day for 5 consecutive days IV | severe | 1B |
| Rituximab | 1g on day 1 and day 14 or 375mg/m2/week for 4 weeks IV | severe | 4 |
| Omalizumab | 300mg every 2-4 weeks SC | severe | 4 |
Levels of evidence: adapted from the Centre for Evidence-Based Medicine, Oxford;
1A: systematic review (with homogeneity) of randomized controlled trials; 1B: individual randomized controlled trials (with narrow confidence interval); 3B: individual case-control study; 4: case series (poor-quality cohort or case-control studies)
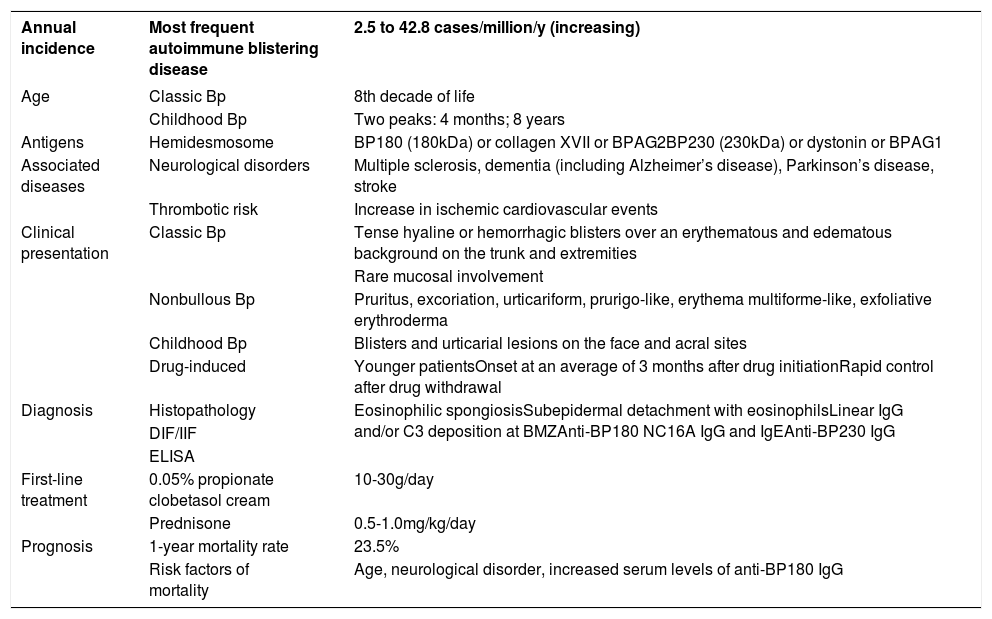
Additional studies are recommended to establish the efficacy and safety of combined therapies in order to reduce the time to achieve disease control and the cumulative dose of topical or systemic steroids without impairing patient’s comorbidities and quality of life. Chart 2 reviews the main BP findings.❑
Bullous pemphigoid in summary
| Annual incidence | Most frequent autoimmune blistering disease | 2.5 to 42.8 cases/million/y (increasing) |
|---|---|---|
| Age | Classic Bp | 8th decade of life |
| Childhood Bp | Two peaks: 4 months; 8 years | |
| Antigens | Hemidesmosome | BP180 (180kDa) or collagen XVII or BPAG2BP230 (230kDa) or dystonin or BPAG1 |
| Associated diseases | Neurological disorders | Multiple sclerosis, dementia (including Alzheimer’s disease), Parkinson’s disease, stroke |
| Thrombotic risk | Increase in ischemic cardiovascular events | |
| Clinical presentation | Classic Bp | Tense hyaline or hemorrhagic blisters over an erythematous and edematous background on the trunk and extremities |
| Rare mucosal involvement | ||
| Nonbullous Bp | Pruritus, excoriation, urticariform, prurigo-like, erythema multiforme-like, exfoliative erythroderma | |
| Childhood Bp | Blisters and urticarial lesions on the face and acral sites | |
| Drug-induced | Younger patientsOnset at an average of 3 months after drug initiationRapid control after drug withdrawal | |
| Diagnosis | Histopathology | Eosinophilic spongiosisSubepidermal detachment with eosinophilsLinear IgG and/or C3 deposition at BMZAnti-BP180 NC16A IgG and IgEAnti-BP230 IgG |
| DIF/IIF | ||
| ELISA | ||
| First-line treatment | 0.05% propionate clobetasol cream | 10-30g/day |
| Prednisone | 0.5-1.0mg/kg/day | |
| Prognosis | 1-year mortality rate | 23.5% |
| Risk factors of mortality | Age, neurological disorder, increased serum levels of anti-BP180 IgG |
Ana Luiza Werneck da Silva, MD, by the Upper Digestive Endoscopy evaluation.
Questions- 1.
Bullous pemphigoid is an autoimmune blistering dermatosis characterized by autoantibodies directed against the following antigens:
- a)
desmogleins 1 and 3
- b)
BP180 and BP230
- c)
desmoplakins I and II
- d)
LABD97 and collagen VII
- a)
- 2.
Which of the following mucocutaneous autoimmune blistering disease is more frequent?
- a)
pemphigus vulgaris
- b)
linear IgA bullous dermatosis
- c)
dermatitis herpetiformis
- d)
bullous pemphigoid
- a)
- 3.
What are the main circulating IgG subclasses in bullous pemphigoid?
- a)
IgG1 and IgG2
- b)
IgG2 and IgG4
- c)
IgG1 and IgG3
- d)
IgG2 and IgG3
- a)
- 4.
In addition to IgG, which other immunoglobulin is commonly involved in the pathogenesis of bullous pemphigoid?
- a)
IgE
- b)
IgA
- c)
IgM
- d)
IgD
- a)
- 5.
What are the main neurological disorders associated with bullous pemphigoid?
- a)
multiple sclerosis, dementia, Parkinson’s disease, epilepsy and cerebrovascular disease
- b)
meningitis, Guillain-Barré syndrome, Alzheimer’s disease, spastic paraparesis and poliomyelitis
- c)
traumatic brain injury, Bell paresis, hydrocephaly, Pick disease and cerebral aneurism
- d)
amyotrophic lateral sclerosis, facial paralysis, Weber syndrome, transverse myelitis and vertebral artery dissection
- a)
- 6.
Frequent locations of bullous pemphigoid lesions are:
- a)
scalp, palmo-plantar and genital region
- b)
arms, buttocks and nails
- c)
face, antecubital and popliteal folds
- d)
axillary region, lower abdomen and inner thighs
- a)
- 7.
The following drugs are inducers of bullous pemphigoid:
- a)
hydroxychloroquine and benzyl penicillin
- b)
furosemide and vildalgliptin
- c)
acetylsalicylic acid and acetaminophen
- d)
omalizumab and rituximab
- a)
- 8.
The main laboratorial findings in bullous pemphigoid are:
- a)
histopathological examination with subcorneal detachment and acantholysis; direct immunofluorescence with intercellular IgG and C3 in the epidermis; indirect immunofluorescence with intercellular IgG
- b)
histopathological examination with suprabasal detachment and acantholysis; direct immunofluorescence with intercellular IgG and C3 in the epidermis; indirect immunofluorescence with intercellular IgG
- c)
histopathological examination with subepidermal detachment with eosinophils and neutrophils; direct immunofluorescence with linear deposits of IgG and/or C3 at the basement membrane zone; indirect immunofluorescence (salt-split skin) with IgG deposition on the epidermal side of the split
- d)
histopathological examination with subepidermal detachment with eosinophils and neutrophils; direct immunofluorescence with linear IgG and/or C3 at the basement membrane zone; indirect immunofluorescence (salt-split skin) with IgG deposition on the dermal side of the split
- a)
- 9.
Risk factors for increased mortality in bullous pemphigoid are:
- a)
blisters over > 80% body surface area; glycemia < 252mg/mL and sodium bicarbonate > 20mmol/L
- b)
underlying malignancy, tachycardia > 120bpm and increased IgE anti-BP180
- c)
older age of onset, neurologic disease, increased IgG anti-BP180 NC16A
- d)
nivolumab therapy, early age of onset, lichenoid lesions
- a)
- 10.
The following medications are recommended in the treatment of bullous pemphigoid:
- a)
intravenous immunoglobulin, colchicine and acitretin
- b)
mycophenolate mofetil, spirolonactone and methotrexate
- c)
azathioprine, isotretinoin and cyclophosphamide
- d)
prednisone, topical clobetasol and dapsone
- a)
Answers
Disseminated leishmaniasis: clinical, pathogenic, and therapeutic aspects. An Bras Dermatol. 2018;94(1):09-16.
1 - D
2 - C
3 - A
4 - C
5 - B
6 - B
7 - D
8 - A
9 - D
10 - D
