








Vasculites são caracterizadas por um processo inflamatório que compromete a parede dos vasos sanguíneos. A gravidade varia desde a existência de casos de mínima repercussão clínica, que comprometem um único órgão, à ocorrência de casos graves com acometimento sistêmico e falência de múltiplos órgãos. Podem ter diversas causas, que determinam, entretanto, padrões histológicos restritos de inflamação vascular. Vasos sanguíneos de qualquer tipo, calibre e de qualquer órgão podem ser afetados, o que resulta em ampla variedade de sinais e sintomas. Vasculites com apresentações clínicas indistinguíveis podem ter prognóstico e tratamento bastante distintos. Essa condição representa um grande desafio para os médicos em termos de classificação, diagnóstico, investigação laboratorial e tratamento, além de caracterizar‐se pela necessidade de acompanhamento cuidadoso. Este artigo revisa a classificação proposta no Consenso Internacional, que ocorreu em Chapel Hill, Carolina do Norte, EUA, em 2012; a etiologia; os recentes conhecimentos obtidos na sua fisiopatologia e especialmente, alerta sobre sintomas e sinais dermatológicos importantes para o diagnóstico de diferentes entidades nosológicas; resume o tratamento de algumas dessas síndromes complexas.
As vasculites englobam grande grupo de doenças heterogêneas caracterizadas por reação inflamatória localizada na parede vascular e nos tecidos perivasculares. Em relação à etiologia, é definida como uma síndrome complexa, pois, embora a maioria seja classificada como idiopática, as vasculites podem ser provocadas por diferentes estímulos. Vasos venosos ou arteriais, ambos podem ser afetados, em um padrão de doença de órgão único ou multissistêmica. Desse modo, lidar com vasculites geralmente significa estudar vários mecanismos fisiopatológicos distintos e um amplo espectro clínico. Muitas vezes, os autores classificam‐nas com base no calibre da parede dos vasos sanguíneos acometidos em: vasculite de vasos pequenos, médios ou grandes. No entanto, quando feita isoladamente, essa classificação falha em reconhecer que vasos do mesmo calibre, ao desempenhar papéis especializados em diversas áreas anatômicas, ao serem afetados podem determinar diferentes padrões de doença.1,2
As vasculites cutâneas podem variar em gravidade, desde erupção cutânea benigna, autolimitada e de curta duração até doença potencialmente fatal com falência múltipla de órgãos. Por sua vez, as vasculites sistêmicas são divididas em duas categorias principais: síndrome da vasculite primária e síndrome da vasculite secundária. A primeira é causada por inflamação dos vasos sanguíneos de etiologia desconhecida. A segunda é induzida por condições subjacentes, inclusive: doenças do tecido conjuntivo, câncer, infecções, imunização e alergia a medicamentos. A maioria dos casos de vasculite cutânea apresenta uma vasculite neutrofílica de pequenos vasos, frequentemente chamada de angeíte leucocitoclástica cutânea (ALC).3
O objetivo desta revisão é esclarecer os conceitos recentes sobre a etiologia das vasculites, a classificação preconizada pela Conferência do Consenso de Chapel Hill e enfatizar as pistas que podem ajudar a sugerir um grupo específico de vasculites na prática clínica durante o exame físico e/ou histopatológico.
Classificação das vasculitesDurante anos, os clínicos julgaram conveniente usar o tamanho do vaso como a característica mais importante para distinguir diferentes formas de vasculites.2 O Consenso de Chapel Hill publicado em 2012 propôs a unificação de novos tipos de síndromes vasculíticas e doenças associadas (tabela 1).4 Entretanto, esse é apenas um sistema de nomenclatura/nosologia, e não uma classificação ou um sistema diagnóstico que ajude a direcionar o manejo clínico das vasculites, pois se baseia eminentemente em agrupá‐las em categorias definíveis, considera determinadas combinações de características.4
Classificação e definições de vasculites adaptadas com base nos critérios adotados pela Conferência do Consenso de Chapel Hill de 2012
| Grupo | Nomenclatura | Definição | Principais características |
|---|---|---|---|
| Vasculites de grandes vasos | Arterite de Takayasu | Arterite, eventualmente granulomas, acometem predominantemente a artéria aorta e seus principais ramos. Início geralmente em pacientes com menos de 50 anos. | Embora artérias de qualquer tamanho possam ser afetadas, acometem grandes artérias com mais frequência do que outras vasculites. As grandes artérias são representadas pela aorta e seus principais ramos. |
| Arterite de células gigantes (ACG) | Arterite, geralmente granulomatosa, acomete a aorta e/ou seus principais ramos, com predileção pelos ramos das artérias carótidas e vertebrais. Muitas vezes envolve a artéria temporal. Início geralmente em pacientes com mais de 50 anos e frequentemente associado à polimialgia reumática. | ||
| Vasculites de médios vasos | Poliarterite nodosa (PAN) | Arterite necrosante de médias ou pequenas artérias, sem glomerulonefrite ou vasculite em arteríolas, capilares ou vênulas, e não associada a ANCAs. | Vasculite afeta predominantemente as artérias de médio calibre, definidas como as principais artérias viscerais e seus ramos. Artérias de qualquer tamanho podem ser afetadas Aneurismas inflamatórios e estenoses são comuns. |
| Doença de Kawasaki | Arterite associada à síndrome do nódulo linfático muco cutâneo, que afeta predominantemente pequenas e médias artérias. Artérias coronárias são frequentemente envolvidas. Aorta e grandes artérias podem estar envolvidas. Geralmente ocorre em bebês ou crianças pequenas. | ||
| Vasculites associadas ao ANCA | Poliangeíte microscópica (PAM) | Vasculite necrosante, com pouco ou nenhum depósito imunológico, afeta predominantemente pequenos vasos (ou seja, capilares, vênulas ou arteríolas). Arterite necrotizante que envolve pequenas e médias artérias pode estar presente. A glomerulonefrite necrosante é muito comum. A capilarite pulmonar frequentemente ocorre. | Vasculite necrotizante, com pouco ou nenhum depósito imunológico (pauci‐imune), afeta predominantemente pequenos vasos (capilares, vênulas, arteríolas e pequenas artérias), associada a ANCA – Mieloperoxidase (MPO) ou Proteinase 3 (PR3) ANCA. Nem todos os pacientes têm positividade para ANCA. Adicione um prefixo que indique reatividade, por exemplo: MPO‐ANCA, PR3‐ANCA, ANCA negativo |
| Granulomatose com poliangeítea (GPA), antiga granulomatose de Wegener | Inflamação granulomatosa necrotizante geralmente envolvendo o trato respiratório superior e inferior, e vasculite necrotizante que afeta predominantemente pequenos e médios vasos (p. ex., capilares, vênulas, arteríolas, artérias e veias). A glomerulonefrite necrotizante é comum. | ||
| Granulomatose eosinofílica com poliangeítea (GEPA), antiga síndrome de Churg‐Strauss | Inflamação granulomatosa necrotizante rica em eosinófilos frequentemente envolve o trato respiratório, e vasculite necrotizante que afeta predominantemente pequenos e médios vasos, associada à asma e eosinofilia. ANCA é mais frequente quando a glomerulonefrite está presente. | ||
| Vasculites por imunocomplexos | Doença anti membrana basal glomerulara (antiga síndrome de Goodpasture) | Vasculite que afeta capilares glomerulares, pulmonares ou ambos, com depósito de autoanticorpos antimembrana basal glomerular. O envolvimento pulmonar causa hemorragia do órgão e o envolvimento renal causa glomerulonefritecom necrose e crescentes. | Vasculite com depósitos moderados a acentuados de imunoglobulina e/ou componentes do complemento na parede dos vasos, que afeta predominantemente pequenos vasos (ou seja, capilares, vênulas, arteríolas e pequenas artérias). A glomerulonefrite é frequente. |
| Vasculite crioglobulinêmica | Vasculite com depósitos de crioglobulinas que afetam pequenos vasos (predominantemente capilares, vênulas ou arteríolas), associados a crioglobulinas séricas. A pele, os glomérulos e os nervos periféricos estão frequentemente envolvidos. | ||
| Vasculite por IgAa (antiga púrpura de Henoch‐Schönlein) | Vasculite, com depósito imune predominante de IgA1, afeta pequenos vasos (predominantemente capilares, vênulas ou arteríolas). Muitas vezes envolve pele e trato gastrointestinal e frequentemente causa artrite. Glomerulonefrite indistinguível da nefropatia por IgA pode ocorrer. | ||
| Urticária‐vasculite hipocomplementêmica(Vasculite Anti‐C1q) | Vasculite acompanhada de urticária e hipocomplementenemia afeta pequenos vasos (ou seja, capilares, vênulas ou arteríolas), associada a anticorpos anti-C1q. Glomerulonefrite, artrite, doença pulmonar obstrutiva e inflamação ocular são comuns. | ||
| Vasculites de vasos de tamanhos variados | Doença de Behçet | Vasculite que pode afetar artérias ou veias. A doença de Behçet é caracterizada por úlceras aftosas recorrentes orais e/ou genitais, acompanhadas por lesões inflamatórias cutâneas, oculares, articulares, gastrointestinais e/ou do sistema nervoso central. Vasculite de pequenos vasos, tromboangeíte, trombose, arterite e aneurismas arteriais podem ocorrer. | Vasculite que pode afetar vasos de qualquer tamanho (pequeno, médio e grande) e tipo (artérias, veias e capilares), sem predominância de um calibre em detrimento de outro. |
| Síndrome de Cogan | Vasculite que ocorre em pacientes com síndrome de Cogan. A Síndrome de Cogan é caracterizada por lesões inflamatórias oculares, inclusive ceratite intersticial, uveíte e episclerite, e doença da orelha interna, inclusive perda auditiva neurossensorial e disfunção vestibular. As manifestações vasculíticas podem incluir arterites (que afetam pequenas, médias ou grandes artérias), aortites, aneurismas aórticos e valvulite aórtica e mitral. | ||
| Vasculites de órgão único | Angeíte cutânea leucocitoclástica | Vasculite cutânea de pequenos vasos | Vasculite em artérias ou veias de qualquer tamanho em um único órgão sem características que indiquem se tratar de uma expressão limitada de vasculite sistêmica. O órgão e o tipo de vaso comprometido devemser incluídosno nome (p. ex.,vasculite dos pequenos vasos cutâneos, arterite testicular,vasculite do sistema nervoso). A distribuição da vasculite pode ser unifocal ou multifocal (difusa) dentro de um órgão.Alguns pacientes originalmente diagnosticados como portadores de vasculite de órgão único desenvolverão manifestações da doença que justificam a redefinição do caso como vasculite sistêmica (p. ex., arterite cutânea que mais tarde se torna PAN sistêmica etc.). |
| Arterite cutânea | Por exemplo, poliarterite nodosa cutânea | ||
| Arterite macular (arterite trombofílica linfocítica) | |||
| Vasculite primária do sistema nervoso central | O diagnóstico de vasculite primária do SNC requer a determinação de que não se trata de um componente de uma outra vasculite (por exemplo, ACG, doença de Behçet, PAM, GPA, GEPA), bem como não está associada à infecção (p. ex., sífilis) ou doença sistêmica (p. ex., LES, sarcoidose) | ||
| Aortite isolada | Aortite idiopática isolada (torácica e/ou abdominal). | ||
| Outras | |||
| Vasculite associada à doença sistêmica | Vasculite lúpica | Vasculite secundária a LES | Vasculite associada e que pode ser secundária a (causada por) uma doença sistêmica. O nome deve ter um prefixo que especifica a doença sistêmica (p. ex., vasculite reumatoide, vasculite lúpica etc.). |
| Vasculite reumatoide | Vasculite secundária à artrite reumatoide | ||
| Vasculite sarcoídea | Vasculite secundária à sarcoidose sistêmica | ||
| Vasculite da policondrite recidivante | Vasculite secundária à policondrite recidivante de múltiplos órgãos | ||
| Outras | Exemplo, relatos de casos de vasculite como manifestação de síndromes autoinflamatórias monogênicas ou poligênicas: síndrome de Behçet autoinflamatória multifatorial e poligênica (pequenos vasos – pele, retina; artéria média – TVP, seio dural, intracraniano, artéria pulmonar. Grande vaso); FMF (Pequenos vasos – púrpura – HSP; tamanho médio – PAN); síndrome de febre periódica associada as criopirinas, CAPS (Pequenos vasos – pele, testículos, rim; relatos de casos raros; HIDS (Pequenos vasos – pele); Armadilhas (Pequenos vasos – pele); síndrome de PAPA (Vasos médios – vasculopatia da artéria cerebral/vasculite); DIRA (vasculite do SNC/vasculopatia), síndrome de Blau (pequenos vasos – retina; vasculite de grandes vasos da pele); DADA2 (inflamação sistêmica, deficiência imunológica leve e vasculopatia manifestada como acidente vascular cerebral recorrente ou PAN). | ||
| Vasculite associada à etiologia provável | Vasculite crioglobulinêmica associada ao VHC | Por exemplo, vasculite crioglobulinêmica de pequenos vasos associada ao HCV. | Vasculite associada a uma etiologia específica provável. O nome(diagnóstico) deve ter um termo de prefixo que especifique a associação (p. ex., poliangeíte microscópica associada à hidralazina, vasculite associada ao VHB, vasculite crioglobulinêmica associada ao HCV etc.). |
| Vasculite associada ao VHB | Por exemplo, poliarterite nodosa associada ao HBV. | ||
| Vasculite associada à sífilis | Vasculite secundária à sífilis | ||
| Vasculite do complexo imune associada ao medicamento | Por exemplo, vasculite do complexo imune associada à doença do soro devido a medicamentos. | ||
| Vasculite ANCA positiva associada a drogas | Por exemplo, poliangeíte microscópica associada à hidralazina, vasculite ANCA positiva associada ao propiltiouracil. | ||
| Vasculite associada ao câncer | Neoplasias hematológicas e de órgãos sólidos, assim como distúrbios linfoproliferativos de células B clonais e síndrome mielodisplásica podem estar associadas e causar vasculite. | ||
| Outras | Vasculite de pequenos vasos devido à doença inflamatória intestinal |
Nova nomenclatura; ANCAs, anticorpos citoplasmáticos antineutrófilos; DADA2, deficiência de adenosina deaminase do tipo 2; DIRA, deficiência do receptor agonista de interleucinas 1; FMF, febre familiar do mediterrâneo; HIDS, síndrome de febre periódica hiper‐IgD; LES, lúpus eritematoso sistêmico; PAN, poliarterite nodosa; PAPA, artrite piogênica, pioderma gangrenoso e acne; PHS, púrpura de Henoch‐Schönlein; TRAPS, síndrome periódica associada ao receptor de fator de necrose tumoral; TVP, tromboembolismo venoso profundo; VHB, vírus da hepatite B; VHC, vírus da hepatite C.
Adaptado de: Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F et al., 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1‐11 e Peleg H, Ben‐Cherit E. Vasculitis in the autoinflammatory diseases. Curr Opin Rheumatol. 2017; 29:4‐11.
Algumas manifestações cutâneas que envolvem pequenos, médios e/ou grandes vasos podem ser encontradas não apenas nas doenças autoimunes, mas também nas doenças autoinflamatórias. Esse termo foi cunhado para descrever uma família emergente de condições caracterizadas por episódios de inflamação não provocada devido à desregulação do sistema imune inato sem o papel principal de linfócitos T autorreativos e/ou autoanticorpos. O envolvimento da pele em doenças autoinflamatórias geralmente se assemelha à vasculite urticariforme ou pode ter características de dermatoses neutrofílicas. As lesões cutâneas podem incluir ainda exantema purpúreo, púrpura não trombocitopênica e pioderma gangrenoso. Em algumas doenças autoinflamatórias, ainda não está claro se a vasculite é uma manifestação clínica integrada ou se representa doença independente adicional.5 Incluímos essas condições em subclassificação apropriada na tabela 1.
Geralmente, o tamanho do vaso acometido tem correlação com a morfologia da lesão observada ao exame dermatológico. O envolvimento de pequenos vasos resulta em: urticária, eritema infiltrado, púrpura palpável ou não, lesões vésico‐bolhosas e/ou pústulas. Úlceras, nódulos, cicatrizes, atrofia branca e livedo racemoso estão associados ao envolvimento dos vasos arteriais musculares, que estão localizados na interface dérmico‐hipodérmica ou no interior do subcutâneo. Em vasculites que envolvem vasos pequenos e médios, todos os tipos de lesões cutâneas descritas anteriormente podem coexistir no mesmo paciente. Finalmente, grande número de úlceras, especialmente quando extensas e associadas à necrose, que pode ser consequência do envolvimento de vasos arteriais profundos, frequentemente prediz vasculite recorrente e doença sistêmica (fig. 1).6
, tipos de vasos sanguíneos encontrados no plexo vascular da derme superficial e profunda, calibre dos casos, local principal de acometimento das síndromes vasculíticas e profundidade adequada da biópsia cutânea para representar a camada da pele onde a doença está localizada.")
Representação esquemática da pele (epiderme, derme e tecido subcutâneo), tipos de vasos sanguíneos encontrados no plexo vascular da derme superficial e profunda, calibre dos casos, local principal de acometimento das síndromes vasculíticas e profundidade adequada da biópsia cutânea para representar a camada da pele onde a doença está localizada.
O reconhecimento de alterações patológicas em biópsia cutânea permite a identificação de diferentes vasculites e sinaliza a possibilidade de progressão de doença cutânea localizada para sistêmica.5 A biópsia de pele é o método padrão‐ouro para o diagnóstico de vasculite cutânea, permite também o estabelecimento de diagnósticos diferenciais, como condições vaso‐oclusivas e outras doenças (tabela 2).
Classificação das vasculites de acordo com o calibre do vaso acometido e achados histopatológicos em amostras de biópsia
| Vasculites de pequenos vasos | Neutrofílicas | Mediadas por imunocomplexos (IFD positiva) | Angeíte leucocitoclástica cutânea (ou vasculite de hipersensibilidade, vasculite cutânea leucocitoclástica ou vasculite cutânea de pequenos vasos) |
| Vasculite por IgA | |||
| Edema agudo hemorrágico da infância | |||
| Urticária‐vasculite | |||
| Vasculites crônicas fibrosantes localizadas: erythema elevatum diutinum, granuloma facial, pseudotumor inflamatório | |||
| Vasculite incidental (IFD negativa) | Síndrome de Sweet e vasculite pustular do dorso das mãos | ||
| Eosinofílicas | Vasculite eosinofílica | ||
| Vasculite eosinofílica cutânea recorrente | |||
| Granulomatosas | Erupções pós‐herpéticas | ||
| Granulomatose linfomatoide | |||
| Linfocíticas | Infecções virais e por Rickettsia | ||
| Dermatose liquenoide, alguns casos, perniose, pitiríase liquenoide, doença do enxerto versus hospedeiro | |||
| Linfomas de células T: linfoma de células T angiocêntrico, papulose linfomatoide | |||
| Reações medicamentosas raras e picadas de artrópodes | |||
| Vasculites mistas, predominantemente de pequenos e médios vasos | Neutrofílicas | Mediadas por imunocomplexos (IFD positiva) | Vasculite crioglobulinêmica associada ao vírus da hepatite C ou paraproteinemia |
| Síndrome da urticária‐vasculite hipocomplementêmica | |||
| Vasculites associadas a doenças do tecido conectivo (ex., lúpus, artrite reumatoide, síndrome de Sjögren) | |||
| Associadas aos ANCAs/pauci‐imunes (IFD negativa) | Granulomatose com poliangeíte | ||
| Granulomatose eosinofílica com poliangeíte | |||
| Poliangeíte microscópica | |||
| Vasculite ANCA positiva induzida por drogas | |||
| Linfocíticas | Doença de Degos | ||
| Infecções virais e por Rickettsia | |||
| Vasculites associadas a doenças do tecido conectivo (ex., síndrome de Sjögren e vasculite lúpica) | |||
| Doença de Behçet | |||
| Miscelânea/Outras | Vasculite séptica | ||
| Doença de Behçet | |||
| Vasculites de médios e grandes vasos musculares | Neutrofílicas | Poliarterite nodosa sistêmica e arterite cutânea | |
| Vasculite Nodular (erythema induratum de Bazin) | |||
| Eosinofílicas | Arterite temporal juvenil | ||
| Granulomatosas | Arterite de células gigantes | ||
| Arterite de Takayasu | |||
| Erupções pós herpéticas | |||
| Linfocíticas | Síndrome de Sneddon | ||
| Doença de Degos | |||
| Doença de Buerger (tromboangeíte obliterante) | |||
| Tromboflebite superficial (Doença de Mondor, linfangite esclerosante) | |||
| Doença de Kawasaki | |||
| Arterite macular (arterite trombofílica linfocítica) |
ANCAs, anticorpos citoplasmáticos anti-neutrófilos; IFD positiva, exame de imunofluorescência direta das lesões da pele mostra depósitos de imunocomplexos na parede dos vasos; IgA, imunoglobulina A.
Adaptado de: Carlson JA, Chen K‐R. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopatol 2006;28(6):486‐506.
Os clínicos precisam ter em mente que a vasculite cutânea, particularmente a vasculite cutânea de pequenos vasos (VCPV), é frequentemente a apresentação mais comum de várias síndromes vasculíticas.1 Ela pode ocorrer em 3% dos casos de granulomatose com poliangeíte (GPA), 13% dos casos de poliangeíte microscópica (PAM) e 28% dos casos de granulomatose eosinofílica com poliangeíte (GEPA).7 Alguns passos são essenciais na prática clínica: (i) anamnese meticulosa e exame físico; (ii) inspeção e palpação das lesões; (iii) biópsia cutânea adequada, de acordo com os achados do exame dermatológico; (iv) exames propedêuticos e complementares, para excluir o envolvimento de órgãos internos; (v) investigação detalhada, inclusive exames laboratoriais para doenças infecciosas e parasitárias, doenças autoimunes do colágeno, neoplasia da medula óssea e/ou câncer de órgão sólido, é útil para avaliação de sua etiologia.
Uma definição importante sobre a nosologia das vasculites na pele foi revista por Sunderkötter et al.1 Os autores enfatizaram que elas podem ser encontradas em cenários clínicos distintos: (i) um componente cutâneo de vasculites sistêmicas: por exemplo, manifestações cutâneas da vasculite por imunoglobulina A (IgA), (ii) uma variante cutânea ou dominante na pele de uma vasculite sistêmica (p. ex., vasculite por IgA limitada à pele) ou (iii) vasculite cutânea de órgão único, que difere em relação aos aspectos clínicos, laboratoriais e características patológicas de vasculite sistêmica reconhecida (p. ex., vasculite nodular). A vasculite cutânea de órgão único nunca evolui para vasculite sistêmica completa; já as formas dominantes da pele podem fazê‐lo. Vasculites sistêmicas, por sua vez, são definidas como aquelas que se apresentam em pelo menos um órgão além da pele.1
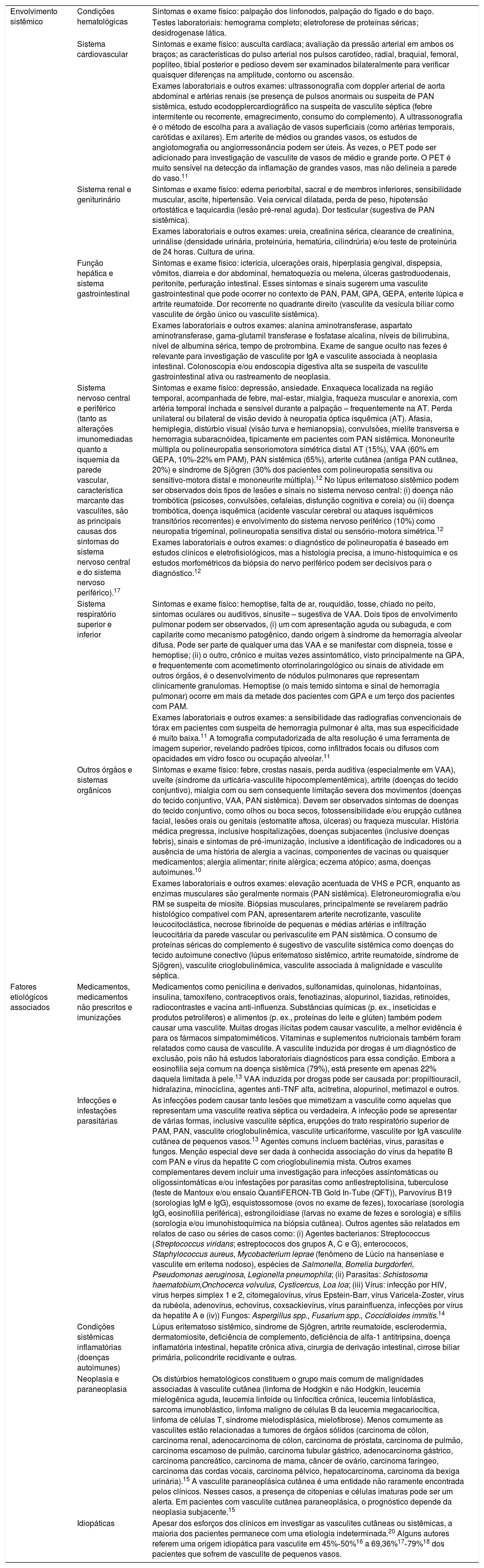
Na maioria dos casos de vasculite cutânea de órgão único, embora possam ocorrer sinais ou sintomas inespecíficos, como leucocitose, aumento da proteína C‐reativa ou artralgia, é improvável manifestação sistêmica significativa.1 Após uma história completa, revisão dos sistemas e exame físico, uma avaliação laboratorial direcionada deve ser feita para confirmar o diagnóstico. Embora não exista um protocolo‐padrão para essa investigação, os testes de rastreamento devem ter como objetivos elucidar uma causa subjacente e avaliar a extensão do envolvimento de outros órgãos, sempre guiados por sinais e sintomas clínicos. Atenção especial deve ser dada a qualquer evidência de vasculite sistêmica, como febre, perda de peso e outros sintomas constitucionais, como artrite; mialgias; dor abdominal, melena ou hematoquezia; tosse, hemoptise ou dispneia; hematúria ou urina espumosa; sinusite ou rinite; e parestesia, fraqueza ou queda do pé. Os médicos também devem perguntar aos pacientes sobre possíveis fatores desencadeantes, inclusive sintomas infecciosos anteriores, uso de medicamentos, vacinas e comorbidades.8–10 Resumimos na tabela 3 os sintomas e/ou sinais de alerta, exames complementares e fatores etiológicos para a investigação das vasculites sistêmicas.11,12
Pesquisa de sinais e sintomas para o diagnóstico de vasculites sistêmicas e exames complementares necessários para identificar possíveis fatores etiológicos associados à doença
| Envolvimento sistêmico | Condições hematológicas | Sintomas e exame físico: palpação dos linfonodos, palpação do fígado e do baço. |
| Testes laboratoriais: hemograma completo; eletroforese de proteínas séricas; desidrogenase lática. | ||
| Sistema cardiovascular | Sintomas e exame físico: ausculta cardíaca; avaliação da pressão arterial em ambos os braços; as características do pulso arterial nos pulsos carotídeo, radial, braquial, femoral, poplíteo, tibial posterior e pedioso devem ser examinados bilateralmente para verificar quaisquer diferenças na amplitude, contorno ou ascensão. | |
| Exames laboratoriais e outros exames: ultrassonografia com doppler arterial de aorta abdominal e artérias renais (se presença de pulsos anormais ou suspeita de PAN sistêmica, estudo ecodopplercardiográfico na suspeita de vasculite séptica (febre intermitente ou recorrente, emagrecimento, consumo do complemento). A ultrassonografia é o método de escolha para a avaliação de vasos superficiais (como artérias temporais, carótidas e axilares). Em arterite de médios ou grandes vasos, os estudos de angiotomografia ou angiorressonância podem ser úteis. Às vezes, o PET pode ser adicionado para investigação de vasculite de vasos de médio e grande porte. O PET é muito sensível na detecção da inflamação de grandes vasos, mas não delineia a parede do vaso.11 | ||
| Sistema renal e geniturinário | Sintomas e exame físico: edema periorbital, sacral e de membros inferiores, sensibilidade muscular, ascite, hipertensão. Veia cervical dilatada, perda de peso, hipotensão ortostática e taquicardia (lesão pré‐renal aguda). Dor testicular (sugestiva de PAN sistêmica). | |
| Exames laboratoriais e outros exames: ureia, creatinina sérica, clearance de creatinina, urinálise (densidade urinária, proteinúria, hematúria, cilindrúria) e/ou teste de proteinúria de 24 horas. Cultura de urina. | ||
| Função hepática e sistema gastrointestinal | Sintomas e exame físico: icterícia, ulcerações orais, hiperplasia gengival, dispepsia, vômitos, diarreia e dor abdominal, hematoquezia ou melena, úlceras gastroduodenais, peritonite, perfuração intestinal. Esses sintomas e sinais sugerem uma vasculite gastrointestinal que pode ocorrer no contexto de PAN, PAM, GPA, GEPA, enterite lúpica e artrite reumatoide. Dor recorrente no quadrante direito (vasculite da vesícula biliar como vasculite de órgão único ou vasculite sistêmica). | |
| Exames laboratoriais e outros exames: alanina aminotransferase, aspartato aminotransferase, gama‐glutamil transferase e fosfatase alcalina, níveis de bilirrubina, nível de albumina sérica, tempo de protrombina. Exame de sangue oculto nas fezes é relevante para investigação de vasculite por IgA e vasculite associada à neoplasia intestinal. Colonoscopia e/ou endoscopia digestiva alta se suspeita de vasculite gastrointestinal ativa ou rastreamento de neoplasia. | ||
| Sistema nervoso central e periférico (tanto as alterações imunomediadas quanto a isquemia da parede vascular, característica marcante das vasculites, são as principais causas dos sintomas do sistema nervoso central e do sistema nervoso periférico).17 | Sintomas e exame físico: depressão, ansiedade. Enxaqueca localizada na região temporal, acompanhada de febre, mal‐estar, mialgia, fraqueza muscular e anorexia, com artéria temporal inchada e sensível durante a palpação – frequentemente na AT. Perda unilateral ou bilateral de visão devido à neuropatia óptica isquêmica (AT). Afasia, hemiplegia, distúrbio visual (visão turva e hemianopsia), convulsões, mielite transversa e hemorragia subaracnóidea, tipicamente em pacientes com PAN sistêmica. Mononeurite múltipla ou polineuropatia sensoriomotora simétrica distal AT (15%), VAA (60% em GEPA, 10%‐22% em PAM), PAN sistêmica (65%), arterite cutânea (antiga PAN cutânea, 20%) e síndrome de Sjögren (30% dos pacientes com polineuropatia sensitiva ou sensitivo‐motora distal e mononeurite múltipla).12 No lúpus eritematoso sistêmico podem ser observados dois tipos de lesões e sinais no sistema nervoso central: (i) doença não trombótica (psicoses, convulsões, cefaleias, disfunção cognitiva e coreia) ou (ii) doença trombótica, doença isquêmica (acidente vascular cerebral ou ataques isquêmicos transitórios recorrentes) e envolvimento do sistema nervoso periférico (10%) como neuropatia trigeminal, polineuropatia sensitiva distal ou sensório‐motora simétrica.12 | |
| Exames laboratoriais e outros exames: o diagnóstico de polineuropatia é baseado em estudos clínicos e eletrofisiológicos, mas a histologia precisa, a imuno‐histoquímica e os estudos morfométricos da biópsia do nervo periférico podem ser decisivos para o diagnóstico.12 | ||
| Sistema respiratório superior e inferior | Sintomas e exame físico: hemoptise, falta de ar, rouquidão, tosse, chiado no peito, sintomas oculares ou auditivos, sinusite – sugestiva de VAA. Dois tipos de envolvimento pulmonar podem ser observados, (i) um com apresentação aguda ou subaguda, e com capilarite como mecanismo patogênico, dando origem à síndrome da hemorragia alveolar difusa. Pode ser parte de qualquer uma das VAA e se manifestar com dispneia, tosse e hemoptise; (ii) o outro, crônico e muitas vezes assintomático, visto principalmente na GPA, e frequentemente com acometimento otorrinolaringológico ou sinais de atividade em outros órgãos, é o desenvolvimento de nódulos pulmonares que representam clinicamente granulomas. Hemoptise (o mais temido sintoma e sinal de hemorragia pulmonar) ocorre em mais da metade dos pacientes com GPA e um terço dos pacientes com PAM. | |
| Exames laboratoriais e outros exames: a sensibilidade das radiografias convencionais de tórax em pacientes com suspeita de hemorragia pulmonar é alta, mas sua especificidade é muito baixa.11 A tomografia computadorizada de alta resolução é uma ferramenta de imagem superior, revelando padrões típicos, como infiltrados focais ou difusos com opacidades em vidro fosco ou ocupação alveolar.11 | ||
| Outros órgãos e sistemas orgânicos | Sintomas e exame físico: febre, crostas nasais, perda auditiva (especialmente em VAA), uveíte (síndrome da urticária‐vasculite hipocomplementêmica), artrite (doenças do tecido conjuntivo), mialgia com ou sem consequente limitação severa dos movimentos (doenças do tecido conjuntivo, VAA, PAN sistêmica). Devem ser observados sintomas de doenças do tecido conjuntivo, como olhos ou boca secos, fotossensibilidade e/ou erupção cutânea facial, lesões orais ou genitais (estomatite aftosa, úlceras) ou fraqueza muscular. História médica pregressa, inclusive hospitalizações, doenças subjacentes (inclusive doenças febris), sinais e sintomas de pré‐imunização, inclusive a identificação de indicadores ou a ausência de uma história de alergia a vacinas, componentes de vacinas ou quaisquer medicamentos; alergia alimentar; rinite alérgica; eczema atópico; asma, doenças autoimunes.10 | |
| Exames laboratoriais e outros exames: elevação acentuada de VHS e PCR, enquanto as enzimas musculares são geralmente normais (PAN sistêmica). Eletroneuromiografia e/ou RM se suspeita de miosite. Biópsias musculares, principalmente se revelarem padrão histológico compatível com PAN, apresentarem arterite necrotizante, vasculite leucocitoclástica, necrose fibrinoide de pequenas e médias artérias e infiltração leucocitária da parede vascular ou perivasculite em PAN sistêmica. O consumo de proteínas séricas do complemento é sugestivo de vasculite sistêmica como doenças do tecido autoimune conectivo (lúpus eritematoso sistêmico, artrite reumatoide, síndrome de Sjögren), vasculite crioglobulinêmica, vasculite associada à malignidade e vasculite séptica. | ||
| Fatores etiológicos associados | Medicamentos, medicamentos não prescritos e imunizações | Medicamentos como penicilina e derivados, sulfonamidas, quinolonas, hidantoínas, insulina, tamoxifeno, contraceptivos orais, fenotiazinas, alopurinol, tiazidas, retinoides, radiocontrastes e vacina anti‐influenza. Substâncias químicas (p. ex., inseticidas e produtos petrolíferos) e alimentos (p. ex., proteínas do leite e glúten) também podem causar uma vasculite. Muitas drogas ilícitas podem causar vasculite, a melhor evidência é para os fármacos simpatomiméticos. Vitaminas e suplementos nutricionais também foram relatados como causa de vasculite. A vasculite induzida por drogas é um diagnóstico de exclusão, pois não há estudos laboratoriais diagnósticos para essa condição. Embora a eosinofilia seja comum na doença sistêmica (79%), está presente em apenas 22% daquela limitada à pele.13 VAA induzida por drogas pode ser causada por: propiltiouracil, hidralazina, minociclina, agentes anti‐TNF alfa, acitretina, alopurinol, metimazol e outros. |
| Infecções e infestações parasitárias | As infecções podem causar tanto lesões que mimetizam a vasculite como aquelas que representam uma vasculite reativa séptica ou verdadeira. A infecção pode se apresentar de várias formas, inclusive vasculite séptica, erupções do trato respiratório superior de PAM, PAN, vasculite crioglobulinêmica, vasculite urticariforme, vasculite por IgA vasculite cutânea de pequenos vasos.13 Agentes comuns incluem bactérias, vírus, parasitas e fungos. Menção especial deve ser dada à conhecida associação do vírus da hepatite B com PAN e vírus da hepatite C com crioglobulinemia mista. Outros exames complementares devem incluir uma investigação para infecções assintomáticas ou oligossintomáticas e/ou infestações por parasitas como antiestreptolisina, tuberculose (teste de Mantoux e/ou ensaio QuantiFERON‐TB Gold In‐Tube (QFT)), Parvovírus B19 (sorologias IgM e IgG), esquistossomose (ovos no exame de fezes), toxocaríase (sorologia IgG, eosinofilia periférica), estrongiloidíase (larvas no exame de fezes e sorologia) e sífilis (sorologia e/ou imunohistoquímica na biópsia cutânea). Outros agentes são relatados em relatos de caso ou séries de casos como: (i) Agentes bacterianos: Streptococcus (Streptococcus viridans; estreptococos dos grupos A, C e G), enterococos, Staphylococcus aureus, Mycobacterium leprae (fenômeno de Lúcio na hanseníase e vasculite em eritema nodoso), espécies de Salmonella, Borrelia burgdorferi, Pseudomonas aeruginosa, Legionella pneumophila; (ii) Parasitas: Schistosoma haematobium,Onchocerca volvulus, Cysticercus, Loa loa; (iii) Vírus: infecção por HIV, vírus herpes simplex 1 e 2, citomegalovírus, vírus Epstein‐Barr, vírus Varicela‐Zoster, vírus da rubéola, adenovírus, echovírus, coxsackievírus, vírus parainfluenza, infecções por vírus da hepatite A e (iv)) Fungos: Aspergillus spp., Fusarium spp., Coccidioides immitis.14 | |
| Condições sistêmicas inflamatórias (doenças autoimunes) | Lúpus eritematoso sistêmico, síndrome de Sjögren, artrite reumatoide, esclerodermia, dermatomiosite, deficiência de complemento, deficiência de alfa‐1 antitripsina, doença inflamatória intestinal, hepatite crônica ativa, cirurgia de derivação intestinal, cirrose biliar primária, policondrite recidivante e outras. | |
| Neoplasia e paraneoplasia | Os distúrbios hematológicos constituem o grupo mais comum de malignidades associadas à vasculite cutânea (linfoma de Hodgkin e não Hodgkin, leucemia mielogênica aguda, leucemia linfoide ou linfocítica crônica, leucemia linfoblástica, sarcoma imunoblástico, linfoma maligno de células B da leucemia megacariocítica, linfoma de células T, síndrome mielodisplásica, mielofibrose). Menos comumente as vasculites estão relacionadas a tumores de órgãos sólidos (carcinoma de cólon, carcinoma renal, adenocarcinoma de cólon, carcinoma de próstata, carcinoma de pulmão, carcinoma escamoso de pulmão, carcinoma tubular gástrico, adenocarcinoma gástrico, carcinoma pancreático, carcinoma de mama, câncer de ovário, carcinoma faríngeo, carcinoma das cordas vocais, carcinoma pélvico, hepatocarcinoma, carcinoma da bexiga urinária).15 A vasculite paraneoplásica cutânea é uma entidade não raramente encontrada pelos clínicos. Nesses casos, a presença de citopenias e células imaturas pode ser um alerta. Em pacientes com vasculite cutânea paraneoplásica, o prognóstico depende da neoplasia subjacente.15 | |
| Idiopáticas | Apesar dos esforços dos clínicos em investigar as vasculites cutâneas ou sistêmicas, a maioria dos pacientes permanece com uma etiologia indeterminada.20 Alguns autores referem uma origem idiopática para vasculite em 45%‐50%16 a 69,36%17‐79%18 dos pacientes que sofrem de vasculite de pequenos vasos. |
ANCA, anticorpos anticitoplasma de neutrófilos; AT, arterite de Takayasu; GEPA, granulomatose eosinofílica com poliangeíte; GPA, granulomatose com poliangeíte; IgA, imunoglobulina A; IgM, imunoglobulina M; IgG, imunoglobulina G; PAM, poliangeíte microscópica; PAN, poliarterite nodosa; PCR, proteína C‐reativa; VAA, vasculites associadas aos ANCAs; VHS, velocidade de hemossedimentação.
Algumas pistas podem ajudar a reconhecer o tamanho do vaso envolvido e, portanto, o diagnóstico suspeito. Hipertensão arterial, por exemplo, pode sugerir vasculite de médios vasos renais, como observado na PAN sistêmica. Lesões cutâneas como púrpura palpável – o tipo de lesão mais frequente na VCPV – púrpura não palpável, pápulas puntiformes, vesículas, pústulas, estrias hemorrágicas subungueais e urticária são frequentemente sugestivas de vasculite de pequenos vasos. Nessa condição, vesículas e bolhas eventualmente surgem sobre máculas purpúricas. Um padrão hemorrágico subsequente é observado e essas lesões podem produzir ulcerações na pele. Púrpura não palpável nas extremidades inferiores pode ser encontrada em pacientes com síndrome de Sjögren e outras condições, que incluem: púrpura hipergamaglobulinêmica, vasculite crioglobulinêmica (VC) e púrpura hipergamaglobulinêmica de Waldenström. Lesões urticariformes podem ocorrer no lúpus eritematoso sistêmico (LES), síndrome de Sjögren, síndrome da urticária‐vasculite hipocomplementêmica e GEPA (fig. 2).13
 Púrpuras, petéquias, vesículas e bolhas hemorrágicas em paciente com vasculite crioglobulinêmica, (b) Lesões urticadas, por vezes confluentes, e púrpuras nos membros inferiores muito sugestivas de urticária vasculite.")
Diferentes lesões normalmente observadas em pacientes com vasculites de pequenos vasos. (a) Púrpuras, petéquias, vesículas e bolhas hemorrágicas em paciente com vasculite crioglobulinêmica, (b) Lesões urticadas, por vezes confluentes, e púrpuras nos membros inferiores muito sugestivas de urticária vasculite.
Em vasculites de vasos de tamanho médio, os pacientes comumente apresentam nódulos subcutâneos, livedo, úlceras grandes ou profundas, lesões papulo‐necróticas e infartos digitais como indicativo de envolvimento oclusivo ou semioclusivo de vasos do plexo da junção dermo‐hipodérmica ou exclusivamente do tecido subcutâneo. Nódulos subcutâneos circundados por padrão livedoide são mais característicos de poliarterite nodosa (PAN), arterite cutânea, vasculite por sarcoidose ou LES do que das vasculites associadas a anticorpos citoplasmáticos (ANCAs) antineutrófilos (VAA) (figs. 3 e 4).13–19
 Nódulos subcutâneos, úlceras e cicatrizes atróficas com bordas hiperpigmentadas em membros inferiores na arterite cutânea, (b) Extensas úlceras com áreas de necrose e cicatrizes atróficas residuais nos membros inferiores em paciente com poliangeíte microscópica.")
Diferentes lesões observadas em pacientes com vasculites de médios vasos: (a) Nódulos subcutâneos, úlceras e cicatrizes atróficas com bordas hiperpigmentadas em membros inferiores na arterite cutânea, (b) Extensas úlceras com áreas de necrose e cicatrizes atróficas residuais nos membros inferiores em paciente com poliangeíte microscópica.
 Livedo racemosa em membros inferiores, inclusive o dorso dos pés em paciente com arterite cutânea, (b) Necroses digitais em doente com vasculite ANCA positiva – granulomatose com poliangeíte.")
As lesões cutâneas dos granulomas de Winkelmann caracterizam‐se pela ocorrência de pápulas ou nódulos sensíveis, eritematosos a violáceos, geralmente simetricamente distribuídos sobre os cotovelos ou extremidades superiores distais e dedos. Raramente foram observadas bandas semelhantes a cordas endurecidas subcutâneas (sinal de corda). Histologicamente, essa doença é uma variante da ALC com influxo proeminente de neutrófilos com macrófagos em paliçada ao redor de uma necrose central composta de fibrina basofílica, colágeno degenerativo e, é claro, neutrófilo e seus restos. Contudo, muitas vezes uma vasculite verdadeira não é observada em seções histológicas. Esse padrão de reação da pele foi originalmente descrito na doença GEPA, mas posteriormente foi observado em uma variedade de doenças infecciosas, como endocardite bacteriana subaguda, hepatite, doenças linfoproliferativas e autoimunes, como GPA, artrite reumatoide, doença inflamatória intestinal, arterite de Takayasu e LES.20
Os infartos digitais são comumente observados na vasculite associada à artrite reumatoide, mas também são vistos na PAN e VAA (fig. 5a).21 Púrpura retiforme em placas (fig. 5b) é um achado incomum de púrpura palpável em padrão livedoide, reticulado ou arciforme. Esse arranjo está relacionado à anatomia vascular fisiológica e resulta de hemorragia relacionada à isquemia ao redor de um vaso dérmico/subcutâneo antes da oclusão completa. A púrpura retiforme em placas pode indicar inflamação vascular, bem como condições não inflamatórias, sem vasculite verdadeira, agrupadas didaticamente como pseudovasculites cutâneas (síndrome antifosfolípide, calcifilaxia, necrose cutânea induzida por warfarina, necrose cutânea induzida por heparina, crioglobulinemia, embolia por colesterol e oxalose).21
: (a) Púrpura palpável com centro necrótico em padrão reticulado livedoide localizada nas coxas. O reconhecimento de um padrão de púrpura retiforme indica a existência de um componente de vasculite em vaso de médio calibre; (b) Ulcerações extensas com bordas irregulares, leito granuloso, presença de áreas de necrose e escara, e descarga serosa.")
Púrpura retiforme em paciente com vasculite ANCA positiva induzida por propiltiouracil (PTU): (a) Púrpura palpável com centro necrótico em padrão reticulado livedoide localizada nas coxas. O reconhecimento de um padrão de púrpura retiforme indica a existência de um componente de vasculite em vaso de médio calibre; (b) Ulcerações extensas com bordas irregulares, leito granuloso, presença de áreas de necrose e escara, e descarga serosa.
Um painel geral de exames complementares sugeridos pelo nosso grupo de estudo inclui vários testes citados anteriormente na tabela 3 e outros, especialmente para aqueles pacientes sem causa óbvia de vasculite, tais como: hemograma, ureia/creatinina séricas, função hepática, urinálise, parasitológico de fezes, eletroforese de proteínas séricas com imunofixação para pesquisa de paraproteinemias, antiestreptolisina O, sorologias para HBV/HCV/HIV, crioglobulinas, níveis de complemento (CH50, C3, C4), anticorpo antinuclear, ANCAs e fator reumatoide (FR).9,13 Se o paciente está febril e tem um sopro cardíaco, então as hemoculturas e a ecocardiografia devem ser feitas. Para casos com envolvimento de vasos de tamanho médio ou sinais e sintomas de doença do tecido conjuntivo, um FAN deve ser obtido.13
Os níveis de proteína C‐reativa geralmente refletem a atividade das VAA. Em ambas VAA e PAN, os títulos de antiestreptolisina O devem ser avaliados devido ao papel comum dos estreptococos nesses cenários. Radiografias de tórax, exames de imagem do sistema gastrointestinal e exame ginecológico/avaliação urológica de acordo com o gênero do paciente são adequados para triagem de malignidades na ausência de quaisquer outros sinais ou sintomas específicos. Febre alta, perda de peso, bicitopenia ou anemia grave, fenômeno de Raynaud (na ausência de doença do tecido conjuntivo) ou crioglobulinas devem levar a uma busca mais completa por uma etiologia neoplásica.13
Uma vez que apenas algumas síndromes vasculíticas têm resultados clínicos, radiográficos e laboratoriais patognomônicos, um diagnóstico confiável e preciso requer uma biópsia de pele representativa para confirmação histológica, cuja interpretação deve ser feita conjuntamente com a história clínica, achados do exame físicos, dados laboratoriais e/ou características angiográficas. Por exemplo, um diagnóstico de vasculite cutânea de órgão único requer que nenhuma evidência de manifestações sistêmicas seja encontrada.10 Se houver suspeita de vasculite sistêmica, estudos de imagem podem ser úteis para determinar a extensão e atividade da doença e exames como proteína C reativa, taxa de sedimentação de eritrócitos e ANCAs devem ser usados para monitorar a atividade da doença e predizer risco de mortalidade.
Assim, a classificação da vasculite cutânea em síndromes específicas é mais bem abordada morfologicamente pela determinação do tamanho do vaso e do principal tipo de células inflamatórias. Esses padrões histológicos (tabela 2), combinados com o exame de imunofluorescência direta (IFD), ANCAs e com os achados de investigação para doenças sistêmicas, permitem diagnóstico específico e, por fim, uma terapia mais eficaz. Uma vasculite de pequenos vasos predominante de neutrófilos que afeta principalmente os vasos dérmicos superiores e mostra depósitos de IgA papilar é diagnóstica de vasculite IgA, enquanto uma vasculite de pequenos vasos predominante de neutrófilos que afeta vasos sanguíneos dérmicos e subcutâneos com predominância de depósitos vasculares de IgM implicaria VC ou vasculite reumatoide.22
Os médicos mais treinados no reconhecimento de lesões cutâneas elementares devem ser capazes de fazer uma biópsia representativa das camadas da pele necessárias. Em geral, deve‐se optar por uma biópsia por punch grande (5‐6 mm de diâmetro) ou excisional.22 Alguns pontos‐chave para uma biópsia cutânea adequada estão listados abaixo:
- a)
Uma amostra de biópsia que se estenda até a tela subcutânea, retirada da pele lesionada, mais avermelhada ou purpúrica, é a chave para se obter um resultado diagnóstico significativo; tempo ideal para a biópsia de pele é < 48h após o aparecimento da lesão. Se a biópsia é mal cronometrada, as características patológicas da vasculite podem estar ausentes, fato que deve ser considerado quando se interpreta uma biópsia negativa de um paciente cujos achados clínicos sugerem vasculite;
- b)
As lesões purpúricas biopsiadas nas primeiras 24 horas exibem depósitos de fibrina dentro da parede do vaso acompanhadas por infiltração neutrofílica da parede e hemorragia circundante e restos nucleares (leucocitoclasia). Após 24 horas, os neutrófilos começam a ser substituídos por linfócitos e macrófagos. Assim, a biópsia de lesões com mais de 48 horas, independentemente da forma subjacente de vasculite, pode mostrar infiltrados ricos em linfócitos e prejudicar o diagnóstico clínico de vasculites sindrômicas;
- c)
As amostras de biópsia devem ser obtidas de lesões não ulceradas, devido ao achado frequente de vasculite incidental subjacente ao leito da úlcera. Se apenas úlceras superficiais estiverem presentes, a biópsia da borda é aceitável. No caso de úlceras profundas, a biópsia do subcutâneo, inclusive a área ulcerada central, aumenta o rendimento diagnóstico e o reconhecimento de uma vasculite arterial, como PAN sistêmica ou arterite cutânea;
- d)
No caso de livedo racemosa, a amostra deve ser retirada do centro do segmento de livedo circular, porque é onde está localizado o vaso estenosado responsável pela periferia cianótica. Nesse cenário, geralmente são necessárias várias seções seriadas para detectar a vasculite, que é tipicamente focal e segmentar.
Mimouni et al.23 revisaram 29 pacientes consecutivos com atrofia branca, 6 dos quais tinham vasculite de tamanho médio subjacente consistente com PAN. Três deles haviam sido previamente diagnosticados como portadores de vasculopatia livedoide em biópsias superficiais. Assim, particularmente no contexto de atrofia branca, biópsias profundas repetidas são frequentemente necessárias para revelar a patologia subjacente. Enquanto isso, Wohlrab et al.24 avaliaram a sensibilidade de biópsias cutâneas na síndrome de Sneddon. Os autores coletaram 5 biópsias com punch profundo (4 mm, no mínimo) de diferentes áreas onde o livedo foi detectado (3 de áreas brancas e 2 de vermelhas) em 15 pacientes. O método teve uma sensibilidade de 27% com uma biópsia, 53% com duas biópsias e 80% com três biópsias retiradas de áreas brancas em todos os casos.
Estudos de IFD devem ser feitos sempre que possível. Por exemplo, o diagnóstico de vasculite por IgA só pode ser feito quando depósitos vasculares de IgA são encontrados.22 Em casos de vasculites mediadas por complexo imune, 100% das amostras de pele revelarão imunoglobulinas nas primeiras 48 horas, ao passo que 70% o farão às 48‐72 horas. Após esse intervalo, apesar de os depósitos do complemento ainda poderem ser detectados em mais de 50% das lesões cutâneas, as imunoglobulinas não serão.22
Nosso grupo fez um estudo retrospectivo sobre os achados à IFD em um centro terciário no Brasil, de janeiro de 2007 a dezembro de 2014. Foram avaliadas 235 amostras cutâneas em 282 pacientes com ALC. A idade variou de 5 a 87 anos, com mediana de 45. Dos pacientes, 191/282 (67,73%) eram do sexo feminino. A análise da IFD mostrou positividade em 70,21% das amostras e C3 foi o imunorreagente mais frequente. A deposição de IgA na parede dos vasos foi relacionada à idade e à ausência de doenças autoimunes/inflamatórias, enquanto a imunoglobulina M (IgM) esteve relacionada ao sexo feminino, distúrbios autoimunes/inflamatórios, consumo de C3 e C4 e positividade anticorpo antinuclear ou anti‐SSA/anti‐SSB. A deposição de imunoglobulina G (IgG) na parede do vaso sanguíneo foi associada à idade e ao ANCA positivo. Finalmente, a deposição de C3 na parede do vaso sanguíneo, o achado mais frequente deste estudo, foi associada com hematúria e envolvimento renal. Envolvimento sistêmico esteve presente em 12,5% dos pacientes.17
Em resumo, nos casos de vasculites cutâneas, é essencial selecionar o local apropriado para a biópsia. As características histopatológicas da ALC podem ser neutrófilos que invadem a parede do vaso, necrose fibrinoide ou fibrina dentro da parede do vaso, poeira nuclear – leucocitoclasia, eritrócitos extravasados, infiltração de células inflamatórias perivasculares, principalmente neutrófilos, e edema endotelial. As biópsias devem ser feitas nas primeiras 8‐24 horas do aparecimento da lesão. Caso contrário, a inflamação destrói os complexos imunológicos e eles se tornam falsamente negativos. A detecção da deposição do complexo imune pode ter significado diagnóstico e prognóstico. Além de imunorreagentes na parede do vaso, a IFD pode revelar a deposição de IgG e C3 na junção dermo‐epidérmica, apontar um teste positivo de banda lúpica, ou pode estar relacionada à vasculite urticariforme hipocomplementêmica com LES subjacente.22
Dados de epidemiologia nas vasculitesA incidência anual de CLA é estimada em 45/1 milhão nos Estados Unidos (população de Olmsted County, Minnesota – casos comprovados por biópsia cutânea).18 Afeta ambos os sexos igualmente e pacientes de todas as idades.9 Em crianças, em contraste a vasculite por IgA é muito mais comum do que a vasculite de pequenos vasos não IgA.9 A presença de uma vasculite sistêmica subjacente, doença do tecido conectivo ou malignidade é muito mais comum em adultos do que em crianças.9,18
Em uma revisão sobre vasculites sistêmicas, Elefante et al.25 apresentaram alguns dados sobre a epidemiologia das VAA: (i) a especificidade do ANCA foi associada a diferentes antecedentes genéticos, características clínicas, resposta ao tratamento e prognóstico em termos de taxas de recidiva e sobrevida; (ii) a distribuição da especificidade do ANCA difere significativamente entre os grupos étnicos; (iii) MPO‐ANCA foi significativamente mais frequente em populações asiáticas [japonês OR = 59,2 (95% IC 8,0–440,7), p < 0,001; chinês OR = 6,8 (95% IC 2,6‐17,8), p < 0,001], branco americano [OR= 2,6 (95% IC 1,7–4,0), p < 0,001] e Oriente Médio/Turquia [OR = 2,3 (95% IC 1,3–4,2), p < 0,005], quando comparados aos europeus do norte, que apresentaram maior especificidade para PR3‐ANCA; (iv) A negatividade da ANCA também foi significativamente mais frequente em americanos brancos do que em europeus do norte [OR = 2,0 (95% IC 1,3–3,2), p = 0,002] e (v) há diferenças na frequência do envolvimento de órgãos entre grupos étnicos. Em comparação com os europeus do norte, a população asiática apresentou significativamente menos envolvimento ocular e de ouvido, nariz e garganta, enquanto o envolvimento renal foi significativamente menos frequente em brancos e significativamente mais frequente no Oriente Médio/Turquia, porém algumas diferenças não foram completamente determinadas pelas diferenças no padrão do ANCA.
PatogênesePodemos simplificar os mecanismos envolvidos na patogênese das vasculites em duas vias: (i) vasculites associadas a complexos imunes e (ii) VAA. No entanto, vários fatores estão envolvidos, como base genética, imunidade inata e adquirida, patógenos, perda da tolerância imunológica e “autoantígenos”.
As causas de VCPV incluem infecções (20%), condições inflamatórias (15%‐20%), reações a medicamentos (10%‐15%) e neoplasias malignas (5%).26 Os fatores indutores ou promotores de doença não são conhecidos em mais da metade dos casos, são atualmente classificados como idiopáticos.10 Embora fatores não imunológicos, como infecção direta das células endoteliais, possam causar vasculite, a maioria das lesões é mediada por mecanismos imunopatogênicos. Esses mecanismos podem ser classificados de acordo com os quatro tipos de reações de hipersensibilidade de Gell e Coombs. A maioria das lesões cutâneas é provavelmente devido à reação de hipersensibilidade tipo III. A deposição de complexos imunológicos em vênulas pós‐capilares ativa o complemento, que, por sua vez, induz a degranulação de mastócitos e a quimiotaxia de neutrófilos.10
Praticamente todos os fármacos têm sido considerados como potenciais agentes precipitantes e a VCPV é de longe a forma mais comum de vasculite associada a medicamentos. No estudo de Ortiz‐Sanjuán et al.,27 os mais comumente associados foram os antibióticos, principalmente B‐lactâmicos e quinolonas. Outros agentes envolvidos foram anti‐inflamatórios não esteroides, paracetamol, alopurinol e anticonvulsivantes. Nessa série de 239 casos, pacientes com vasculite devido a infecções importantes, como endocardite ou pneumonia, foram excluídos.
A vasculite reumatoide é provavelmente a forma mais amplamente reconhecida de vasculite secundária e é tipicamente observada na artrite reumatoide soropositiva, erosiva e de longa duração. Acredita‐se que a deposição de complexos imunes e subsequente ativação da cascata do complemento desempenhe um papel fundamental em sua patogênese. Uma expressão aumentada de citocinas inflamatórias, como TNF‐α, IL‐1 e IL‐6, é notada, embora seu papel exato na fisiopatologia da doença ainda não esteja claro. Vasculite cutânea pode ser observada em aproximadamente 19%‐28% dos pacientes com LES.28 Pequenos vasos, especialmente vênulas pós‐capilares, estão envolvidos na maioria dos casos (80%‐90%).28 Presença de anticorpo antiendotelial é detectada em até 80% desses pacientes e pode contribuir por meio da ativação de células endoteliais, efeito citotóxico direto devido à citotoxicidade dependente de complemento ou efeito citotóxico indireto secundário à citotoxicidade dependente de anticorpos.28
Suscetibilidade genética para vasculitesEstudos de associação genômica ampla revelaram um papel central da região do antígeno leucocitário humano (HLA) na suscetibilidade genética às vasculites. No entanto, cada forma tem marcadores distintos de HLA, provavelmente devido a diferenças específicas em termos de fatores antigênicos. Apesar do avanço considerável na identificação de fatores de risco genéticos durante os últimos 10 anos, o número de locus de risco identificados para a maioria dos tipos de vasculites permanece significativamente menor do que aqueles identificados para outras doenças imunomediadas.29
Há uma forte associação dos HLA de classe II com a patogênese da vasculite por IgA, especificamente HLA‐DRB1 em europeus, principalmente devido ao HLA‐DR1*0103.43, sugere que possa estar relacionada a outras vasculites de classe II, como arterite de células gigantes ou VAA. Também foi proposto que, embora menos forte, há um efeito potencial do HLA classe I nessa vasculite. Quando se trata das VAA, as distinções genéticas dentro do grupo podem estar relacionadas à especificidade antigênica do ANCA, e não à síndrome clínica. Diferentes subtipos de VAA são sustentados por fatores de risco genético distintos, a GPA é associada ao HLA‐DP, SERPINA1, PRTN3 e semaforina 6A, ao passo que a PAM está principalmente associada a polimorfismos do HLA‐DQ.29–31
A arterite de Takayasu é predominantemente relacionada aos alelos HLA classe I, em particular ao HLA‐B52, assim como a doença de Behçet.30 O fator de risco isolado mais importante para a doença de Behçet foi confirmado como o HLA‐B51, mas outro alelos HLA‐B com menor efeito independente também foram observados, por exemplo, HLA‐B15 e HLA‐B27.29 Fora da região do complexo de histocompatibilidade principal, a suscetibilidade à doença tem sido associada à interleucina 10 e à região do receptor da interleucina 23/β2da interleucina 12.29
A associação de alelos HLA‐DR com PAN foi relatada, mas com resultados variáveis de acordo com a população estudada. Infelizmente, há pouca informação sobre a suscetibilidade genética à vasculite de hipersensibilidade (VCPV ou vasculite cutânea de órgão único). Estudos genéticos sobre essa condição são escassos e nenhuma associação com a região HLA foi confirmada, sugere uma etiologia heterogênea na patogênese dessa vasculite que muitas vezes é restrita e limitada à pele.30
Infecções como gatilho de vasculitesVários mecanismos imunes estão envolvidos na patogênese das vasculites associadas à infecção. A maioria deles é causada por invasão direta e proliferação de patógenos nas paredes vasculares, gera inflamação. Por outro lado, às vezes podem ser atribuídos a efeitos imunológicos indiretos, como reações de hipersensibilidade tipo II, III ou IV, desencadeadas pela infecção. Estruturas como fímbrias, além de MSCRAMMs (componentes de superfícies microbianas que reconhecem moléculas de matriz adesivas) e outras proteínas ancoradas na parede celular, são usadas pelas bactérias para chegar às células vulneráveis. Foi demonstrado que os vírus têm moléculas de adesão de membrana análogas. No entanto, o conhecimento se a sua densidade de expressão tem um papel na determinação da frequência com a qual órgãos distintos são afetados precisa ser buscado em pesquisas científicas futuras.2,32
Vasculite foi relatada em associação com numerosos microrganismos, desde vírus a fungos. Em situações específicas, a relação entre etiologia e inflamação/destruição angiocêntrica é clara, como ocorre na aortite por Mycobacterium tuberculosis ou Treponema pallidum, nas quais há uma predileção pela aorta ascendente. No entanto, outras infecções, como o HIV, estão associadas a uma ampla variedade de fenótipos, afetam vasos pequenos, médios ou grandes. Nesses casos, diversos padrões, como arterite necrosante, não necrosante, de células gigantes e eosinofílica, foram observados na histologia. Invasão direta da parede do vaso pelo próprio HIV ou por organismos oportunistas, bem como complexos imunes com antígenos e anticorpos do HIV, são alguns dos mecanismos de lesão propostos.33
De fato, várias doenças infecciosas estão implicadas na produção de vasculites mediadas por complexos imune.2 Foi relatado que a vasculite por IgA ocorre em associação com várias bactérias, inclusive Mycoplasma pneumoniae e Clostridium difficile. Ureaplasma urealyticum, Aerococcus viridians, Burkholderia cepacia e Listeria monocytogenes foram incriminadas na ALC.33 Embora a incidência da PAN tenha diminuído significativamente com o avanço dos programas de vacinação e triagem de transfusões sanguíneas, mais de 30% dos casos são secundários à infecção por HBV. A maioria das evidências corrobora uma reação de hipersensibilidade do tipo III, em que o dano vascular é produzido por complexos imunes com antígenos virais. Tem havido relatos anedóticos de infecção crônica por HBV associada a outras formas de vasculite, como a GPA, embora ainda não esteja claro se esses relatos são causais ou coincidentes.32
A patogênese da vasculite associada ao HCV parece envolver uma interação direta entre o vírus e os linfócitos, leva à ativação policlonal e à proliferação de células B, produz IgM com atividade de FR.33 O FR IgM é capaz de ativar o sistema do complemento através da ligação do domínio globular da proteína C1q. Os receptores C1q (gC1q‐R) são amplamente expressos na superfície das células sanguíneas e células endoteliais e se ligam a grandes imunocomplexos com proteínas do núcleo do HCV, facilitam a inflamação vascular subsequente. Os imunocomplexos com crioprecipitado que contêm antígenos virais, FR IgM ligado à IgG policlonal anti‐HCV e complemento são localizados em pequenos vasos de órgãos internos, embora preferencialmente ocorram nas extremidades mais frias.32
A VC associada ao VHC é uma vasculite sistêmica de vasos pequenos a médios, devido à deposição vascular de proteínas séricas precipitáveis ao frio, denominadas crioglobulinas. As crioglobulinas do tipo I são imunoglobulinas monoclonais, as crioglobulinas tipo II consistem em imunoglobulinas monoclonais com atividade de FR, associadas à IgG policlonal, enquanto as crioglobulinas tipo III compreendem IgG e IgM policlonais com atividade de FR. Elefante et al.25 resumiram os fatores clínicos, laboratoriais e predisponentes que influenciam o prognóstico em pacientes com VC não relacionada ao HVC: (i) essencial (doença subjacente não identificável): VC foi o maior grupo (39,4%) e a primeira condição associada (21,1%) foi síndrome de Sjögren primária (SSp); (ii) púrpura esteve presente em 78% dos pacientes do grupo pSS, 64% dos quais apresentaram crioglobulinas tipo II, e em pacientes com SSp a presença de crioglobulinas foi associada com maior atividade sistêmica, sugeriu que todos esses pacientes devem ser testados para crioglobulinas séricas, pelo menos no momento do diagnóstico; (iii) VC relacionada ao LES esteve presente em 10,9% dos casos, assim como outras condições imunológicas, positividade HBsAg em 8,6%, doenças linfoproliferativas em 6,8% e alguns tumores sólidos em 2,3%; (iv) crioglobulinas tipo II foram detectadas em 54,9% e estavam independentemente associadas à púrpura e à fadiga, e (v) idade avançada, sexo masculino, crioglobulinas tipo II e HBsAg foram independentemente associados com grande mortalidade em pacientes com VC não relacionada ao HCV.
A PAN é dividida em dois subtipos: sistêmico e cutâneo. Apresenta‐se predominantemente entre os 40 e os 60 anos. Além do VHB, a PAN sistêmica esteve associada a outras condições infeciosas, como HCV, HIV, citomegalovírus, parvovírus B19 e HTLV. A PAN cutânea tem sido atribuída a infecções estreptocócicas na população pediátrica.26
Vasculites associadas ao ANCAOs ANCAs são um grupo de autoanticorpos, predominantemente do tipo IgG, desenvolvidos e liberados por células B contra antígenos no citoplasma dos neutrófilos granulócitos. Um tipo de ANCA está associado à coloração difusa do citoplasma e é conhecido como ANCA citoplasmático (c‐ANCA), enquanto o outro está relacionado com a coloração ao redor do núcleo e é conhecido como ANCA perinuclear (p‐ANCA). O principal antígeno visado pelo c‐ANCA é PR3 e o alvo de p‐ANCA é MPO. Ambos são expressos na superfície celular de neutrófilos ativados por citocinas pró‐inflamatórias, como IL‐1β e TNF gerados durante infecções. Os ANCAs ligam‐se a esses antígenos. Entretanto, sua porção do fragmento cristalizável (Fc) liga‐se aos receptores Fc nos neutrófilos, induz sua ativação excessiva. Esse fato leva à produção anormal de citocinas, liberação de espécies reativas de oxigênio (ROS) e enzimas líticas, e, eventualmente, à formação de armadilhas extracelulares de neutrófilos (NET).34,35
As VAA representam um grupo de doenças raras caracterizadas por inflamação necrosante de vasos sanguíneos pequenos ou médios, com a presença de ANCAs. São elas: GPA, MPA com a sua forma limitada renal – glomerulonefrite necrotizante crescêntica pauci‐imune e GEPA. Resultados de estudos demonstraram que os ANCAs, detectados pela técnica de imunofluorescência (c‐ANCA ou p‐ANCA), são um marcador sensível para as chamadas VAA, a sensibilidade varia de 80% a mais de 90%. Infelizmente, a imunofluorescência tem uma baixa especificidade (80% ou menos), que está relacionada principalmente à positividade de p‐ANCA nos controles portadores de doenças inflamatórias intestinais. O p‐ANCA nos controles também pode ser causado pela presença de anticorpos antinucleares. A especificidade antigênica (PR3 ou MPO) não diferencia de forma efetiva as VAA, porém o c‐ANCA/PR3‐ANCA é encontrado principalmente na GPA, enquanto o p‐ANCA/MPO‐ANCA é mais prevalente em PAM, glomerulonefrite necrotizante crescêntica pauci‐imune e GEPA. Os ANCAs são detectados em 70%‐90% dos casos de GPA ativa generalizada, mas apenas em cerca de 40%‐50% das formas locorregionais.36 Além de um marcador diagnóstico, os dados reiteram o papel patogênico dos ANCAs, que promovem a ativação de neutrófilos estimulados e monócitos e sua adesão ao endotélio, levam a um dano tecidual subsequente.19
As proteínas do sistema complemento participam da patogênese de diversas vasculites de pequenos e médios vasos, inclusive VAA, VC, vasculite por IgA e vasculite urticariforme. Neutrófilos ativados por PR3 e MPO‐ANCA estimulam a liberação de C5a (um derivado proteico da ativação da via alternativa do complemento).30 A interação subsequente de C5a com o receptor C5a 1 (C5aR1) pode representar uma alça de amplificação pró‐inflamatória nas VAA. Devido a essa ativação do complemento, foram observadas concentrações séricas e plasmáticas elevadas de C5a e C3a nas VAA ativas, especialmente naquelas MPO‐ANCA positivas.31
Além de componentes microbianos, como peptídeos ou lipopolissacarídeos e complemento, certas drogas, principalmente propiltiouracil, hidralazina e cocaína adulterada com levamisol, estimulam a expressão dos ANCAs na superfície da membrana de neutrófilos. Embora essas células imunes sejam o ator principal na patogênese das VAA, uma vez que são células efetoras responsáveis por danos endoteliais e alvos de autoimunidade, foi demonstrado que outras células, inclusive células T CD4 e monócitos, também estão envolvidas.35
Armadilhas extracelulares de neutrófilos (NETs) e vasculitesOs neutrófilos são capazes de gerar NETs, que são encontradas em uma variedade de condições, como infecções, malignidades, aterosclerose e, como mencionado anteriormente, doenças autoimunes, como VAA. Embora vários estímulos sejam responsáveis pela ativação de neutrófilos e formação de NETs, pouco se sabe sobre a via exata pela qual isso ocorre.37 A mais forte evidência de que as NETs realmente servem como uma fonte de autoantígenos que estimulam a produção de autoanticorpos nas vasculites vem de estudos sobre vasculites induzidas por drogas. A positividade de MPO‐ANCA é relativamente comum em pacientes tratados com propiltiouracil e alguns deles desenvolvem uma síndrome semelhante à vasculite.38 MPO e PR3 foram demonstrados dentro de NETs isoladas de pacientes com VAA e, por sua vez, os ANCAs podem induzir ainda mais a formação de NETs. Os ANCAs também podem induzir a autofagia de neutrófilos, o que promove a NETosis, uma forma única de morte celular programada.38
As NETs mostraram estar presentes não apenas em lesões de pele e em trombos de pacientes com VAA, mas também na circulação. Acredita‐se que as NET com proteínas pró‐inflamatórias contribuem para a vasculite por dano direto às células endoteliais, ativação do sistema complemento (especialmente via alternativa do complemento), geram p/c‐ANCA e trombose.37 No entanto, Wang et al.39 demonstraram que os níveis circulantes de NETs não podem ser usados como um biomarcador para avaliar a atividade da doença nesses pacientes.
ConclusãoGrande número de contribuições significativas foi feito sobre a classificação, patogênese, subdefinição clínica de vasculites cutâneas e sistêmicas neste trabalho. A principal razão para a pesquisa em andamento é buscar medicina personalizada baseada em endótipos, em vez de fenótipos. A capacidade de reconhecer lesões tegumentares de diferentes vasculites, aliada a outros sintomas e sinais sistêmicos, biópsia e exames laboratoriais e de imagem adequados, pode contribuir para o correto diagnóstico desse desafiador grupo de doenças.
Suporte financeiroCoordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), Código de Financiamento 001.
Contribuição dos autoresThamara Cristiane Alves Batista Morita: Elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito.
Gabriela Franco S. Trés: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Roberta Fachini Jardim Criado: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; revisão crítica do manuscrito.
Mirian Nacagami Sotto: Revisão crítica do manuscrito.
Paulo Ricardo Criado: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Como citar este artigo: Morita TCAB, Trés GFS, Criado RFJ, Sotto MN, Criado PR. Update on vasculitis: an overview and dermatological clues for clinical and histopathological diagnosis ‐ Part I. An Bras Dermatol. 2020;95:355–71.
Trabalho realizado no Departamento de Dermatologia, Faculdade de Medicina, Universidade de São Paulo e no Departamento de Dermatologia, Faculdade de Medicina do ABC, Santo André, SP, Brasil.
