



Tuberous sclerosis complex is a multisystemic, autosomal dominant genetic disorder with complete penetrance, that can evolve with hamartomas in multiple organs, such as skin, central nervous system, kidney and lung. Due to the wide phenotypic variability, the disease is often not recognized. Tuberous sclerosis complex affects one in 10,000 newborns and most patients are diagnosed during the first 15 months of life. The diagnostic criteria for tuberous sclerosis were reviewed in 2012, at the second International Tuberous Sclerosis Complex Consensus Conference. The diagnosis is based on genetic criteria, by the identification of inactivating pathogenic mutation of tumor suppressor genes TSC1 and TSC2, and clinical criteria, including cutaneous, renal, pulmonary, cardiac and neurological manifestations. The treatment of tuberous sclerosis complex consists, mainly, in management of the symptoms caused by hamartomas and in prevention of organ failure. Multidisciplinary approach is recommended, in order to obtain better clinical outcomes.
Tuberous sclerosis complex (TSC) is a multisystemic neurocutaneous genetic condition with autosomal dominant inheritance, characterized by hamartomas that affect multiple organs, including skin, central nervous system, heart, lungs, and kidney. It is also known as epiloia or Pringle-Bourneville phacomatosis, and was initially described in the 19th century by Virchow and Von Recklinghausen, who identified hamartomas in the brain and heart during the necropsy of patients with seizures and mental retardation. However, the correlation between the cutaneous manifestations with other clinical symptoms and the description of the syndrome were made by Bourneville in the beginning of the 20th century. Years after, Campbell in 1905 and Vogt three years later established the triad that characterizes TSC, which is mental retardation, epilepsy and Pringle type of sebaceous adenoma (angiofibroma).1,2
Diagnostic criteria for tuberous sclerosis were firstly established in 1998.3 In 2012, in the second International Tuberous Sclerosis Complex Consensus Conference held in Washington, these criteria were reviewed with the aim of presenting recommendations for the diagnosis, surveillance and management of TSC patients.3
The condition affects one in every 6,000 to 10,000 individuals and can affect both sexes and all ethnic groups equally. It has a great phenotypical variability, which can sometimes make its recognition difficult.4,5
Epidemiology and EtiopathogenesisTSC occurs due to the deletion, rearrangement and inactivating mutation of tumor suppressor genes TSC1 or TSC2, that lead to abnormal proteins hamartin and tuberin, codified in the loci 9p34 and 16p13, respectively.4,6 The role of these genes consists in the regulation of cellular growth through the phosphatidylinositol 3-kinase signaling pathway, inhibiting the mammalian target of rapamycin (mTOR).7 The complex hamartin/tuberin is an important inhibitor of tumor growth. These proteins suppress the activity of the mTOR pathway, responsible for cellular proliferation and inhibition of cellular apoptosis. In TSC patients, changes in these proteins lead to a permanent activation of the mTOR pathway, and therefore to the formation of hamartomas in multiple organs. Familial cases of the condition are due to germline mutations and, despite being able to be transmitted hereditarily, 70% of TSC patients are the result of somatic mutations, configuring sporadic cases.1 Studies demonstrate that changes in the TSC2 gene are more common and lead to a more severe neurological impairment when compared to TSC due to mutations in TSC1.5,8,9 Cases of familial transmission result in mild to moderate disease, sometimes not fulfilling all diagnostic criteria and have a higher frequency of changes in the TSC1 gene.
Clinical ManifestationsMost patients affected by TSC seek medical attention due to seizures or skin lesions.10 TSC is a condition with variable expression and complete penetrance. Both sexes are equally affected but women can have more marked signs.1 Cutaneous manifestations represent the most common findings observed in TSC patients, even though there are some affected individuals with no cutaneous involvement. Neurological and renal complications are the main cause of morbidity and mortality associated to the condition.
Cutaneous manifestationsSkin lesions are detected in all ages and affect more than 90% of TSC patients.10
Among the cutaneous manifestations, hypopigmented macules are present in 90% of the cases and are usually found at birth, making it possible to suspect the diagnosis at an early age. When the cutaneous lesions are associated to other clinical findings, the definitive and early diagnosis of the syndrome can be made.3,5,6 The most typical macules are hypopigmented and have a leaf shape, round in one end and pointed in the other, the so called “ash-leaf”. They are usually larger than 5mm. Over the years, new macules can appear, including smaller, round hypopigmented macules known as “confetti” lesions. In adulthood, “ash-leaf” lesions tend to become milder, with repigmentation and sometimes they can disappear. Clinically, “confetti” lesions and guttate leukoderma are difficult to differentiate, however, the former appear during childhood and can be asymmetrically distributed over the body.1
Facial angiofibromas are observed in 83 to 90% of cases and usually appear in the first decade of life, around the 3rd or 4th year of life. The number of lesions can increase during adolescence, becoming stable during adulthood. They are characterized by papules that affect mainly the malar, nasal dorsum, nasolabial fold, forehead and chin areas, ranging in color from normal, when they have more fibrous tissue, to violaceous, where blood vessels predominate.4,11 Even though they are usually asymptomatic, angiofibromas can bleed spontaneously, impair eye sight and affect the quality of life due to the aesthetic disfiguration.12 In some patients, these lesions become confluent, causing marked disfiguration.13
Connective tissue nevi are normal-colored or brown plaques that can be present at birth or appear during childhood and adolescence. Among these lesions, some are called shagreen patches when located on the lumbosacral region, with the orange peel appearance and histologic features similar to other collagenomas found on the trunk. Fibrous cephalic plaque, considered the most specific TSC lesion, is present in 25% of cases, and is more commonly found on one side of the forehead but it can also be seen in other areas of the face and scalp.1,3,14
Ungual fibroma or Koenen tumor are tumors observed later, usually after the second decade of life and can progressively increase in size. It has predilection for females and for the toes and generally can cause longitudinal ridging on the nails when they affect the ungual matrix.1
In the oral cavity, the most common lesions are pits on the teeth enamel and gingival fibromas. Dental pits are more common in TSC patients compared to the general population and need a through clinical examination for their diagnosis, being not seen many of the times. Gingival fibromas are seen in 20 to 50% of the patients, most during adulthood, and can be normal-colored or violaceous, usually on the anterior gingiva.3
Renal manifestationsRenal angiomyolipomas are extremely common benign tumors in TSC patients, observed in 80% of cases. They progressively enlarge from the first years of life, with an obvious growth during adolescence or early adulthood and stop enlarging in the elderly in about 30% of cases.15,16 They are responsible for a marked morbidity and mortality in TSC patients, despite most of them being asymptomatic. When there are clinical manifestations from these tumors, hematuria and loss of kidney function are highlighted, as well as risk of hemorrhage for tumors larger than 30mm. Besides angiomyolipomas, TSC patients have a higher incidence of multiple renal cysts when compared to the general population, due to the mutation in the TSC1 and TSC2 genes.15,16
There are reports of TSC associated to the renal cell carcinoma in children and young adults, considered to be a rare manifestation, with an overall incidence of around 1 to 2%, what makes the diagnosis challenging. Many times, it is difficult to distinguish it from angiomyolipomas with sparse fat tissue through conventional imaging studies. If this diagnosis is suspected, a biopsy with histology is recommended to avoid invasive surgical procedures. There is little information in the literature about its prognosis and response to conventional treatments.17,18
Pulmonary manifestationsReplacement of alveolar tissue by cysts and proliferation of the smooth muscle, known as lymphangioleiomyomatosis, progressively affect the pulmonary function in 40% of the patients.15 It affects mainly adult females, being rare the symptomatic cases in men. The most common signs and symptoms are cough, dyspnea, hemoptysis and pneumothorax.
Cardiac manifestationsRhabdomyoma is the most frequent cardiac tumor, appearing early in the fetus and newborn, around between the 20th to 30th weeks of gestation, more commonly seen in the ventricle wall.8 The repercussions of these tumors vary according to the location and number of lesions. Most of the time they can be asymptomatic, or evolve with cardiomegaly, murmurs, changes in the blood flow, arrhythmias, non-immune hydrops and death. Among breastfeeding babies with multiple cardiac rhabdomyomas, 80% or more have TSC. These lesions can be identified from 22nd week of gestation. Most patients have an average of three lesions, measuring approximately 3 to 25mm in size but isolated lesions are also of diagnostic importance.5 In the clinical course, these tumors usually involve completely within the first years of life.1
Neurological manifestationsThe main neurological manifestation of TSC is epilepsy, which occurs in 70 to 90% of the individuals with the condition and this is the sign that most frequently leads to the diagnosis of the syndrome. Seizures usually start within the first three years of life, typically as infantile spasms and focal seizures. However, it is possible to find all types of seizures in TSC patients and in two thirds of the cases there is initially focal refractory epilepsy.19 Moreover, these patients present a higher risk for other neurocognitive deficits such as manifestations in the autism spectrum, mental retardation and mood disturbances.15 Neuroimaging studies can show cortical and subcortical tubers and migration lines in the white matter, these being the manifestations responsible for cortical dysplasia.19 Other findings include subependymal nodules in the wall of the lateral ventricles and a third ventricle in 80% of the cases that are usually asymptomatic; and also giant cell subependymal astrocytomas, observed in 5 to 15% of TSC patients, which in turn can lead to ventriculomegaly and hydrocephalus, resulting in marked morbidity and mortality.1,19 The disturbances in the autism spectrum, among other behavioral problems, can be identified in 40% to 50% of the patients. These have a prevalence of 75% of coexisting cognitive impairment and an additional prevalence of 75% to 100% of concomitant epilepsy.5 In addition, the term TAND (tuberous sclerosis-associated neuropsychiatric disorders) was introduced to illustrate all the biopsychosocial difficulties seen in patients with tuberous sclerosis.19 Some of them include behavior changes, aggressiveness, anxiety, depressive mood, sleep disorders, and learning difficulties. Changes in the neurodevelopment such as attention deficit and hyperactivity are found in 30% to 40% of TSC patients.19
Ophthalmologic abnormalitiesRetinal hamartoma can be present in 30% to 50% of patients and tends to be bilateral and multiple, becoming apparent during infancy. In the majority of patients, the lesions do not cause visual impairment. Patients can also present hypopigmented macule in the retina, which corresponds to the hypopigmented macules seen on the skin.
Liver abnormalitiesGastrointestinal manifestations are uncommon in TSC patients and rarely liver angiomyolipomas can be found, which have a predilection for females.3,20
Diagnosis and Follow-UpThe diagnostic criteria for tuberous sclerosis were reviewed in 2012, in the second International Tuberous Sclerosis Complex Consensus Conference, after a meeting with 79 experts from 14 countries, with the objective of establishing recommendations for the diagnosis, surveillance and management of TSC patients. Some changes were made comparing to the previous criteria (1998),3 being the most relevants the inclusion of genetic testing, the refinement of the clinical criteria and the exclusion of probable diagnosis.3
According to the new genetic criterion, the identification of the pathogenic mutation in TSC1 or TSC2 DNA is enough to establish the definitive diagnosis of TSC, configuring an independent diagnostic criterion. Molecular testing yields a positive result in 75% to 90% of TSC patients; in a significant fraction (10% to 25%), conventional genetic testing did not identify the pathogenic mutation. Therefore, a normal result in the molecular testing does not rule out TSC. However, if the mutation is identified in an affected patient, genetic testing has a high predictive value for the other family members.3 The growing availability of molecular testing and the advent of sophisticated technologies allowed genetics to become a great contributor for the diagnosis of TSC. This new criterion facilitates diagnosis, especially in breast-feeding babies who still have not fulfilled the diagnostic criteria based on clinical manifestations. In countries where the analysis of the mutations is less available, clinical criteria continue to be the main way of establishing the diagnosis of tuberous sclerosis.3,19
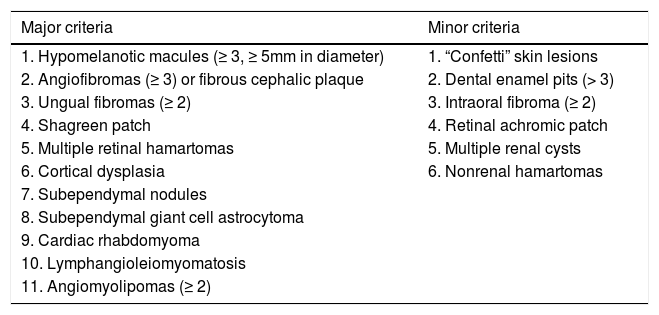
Clinical criteria are divided into major: hypopigmented macules (≥ 3, with at least 5mm diameter), angiofibromas (≥ 3) or fibrous cephalic plaque, ungual fibromas (≥ 2), shagreen patch, multiple retinal hamartomas, cortical dysplasia, subependymal nodules, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis, angiomyolipomas (≥ 2); and minor: “confetti” lesion, enamel pits (> 3), intraoral fibroma (≥ 2), retinal hypopigmented macule, multiple renal cysts and nonrenal hamartomas (Chart 1, Figure 1 and Figure 2).3 The definitive diagnosis is defined by the presence of two major criteria or one major and two minor criteria. Probable diagnosis is made with one major criteria or two or more minor.
Clinical criteria for the diagnosis of tuberous sclerosis complex
| Major criteria | Minor criteria |
|---|---|
| 1. Hypomelanotic macules (≥ 3, ≥ 5mm in diameter) | 1. “Confetti” skin lesions |
| 2. Angiofibromas (≥ 3) or fibrous cephalic plaque | 2. Dental enamel pits (> 3) |
| 3. Ungual fibromas (≥ 2) | 3. Intraoral fibroma (≥ 2) |
| 4. Shagreen patch | 4. Retinal achromic patch |
| 5. Multiple retinal hamartomas | 5. Multiple renal cysts |
| 6. Cortical dysplasia | 6. Nonrenal hamartomas |
| 7. Subependymal nodules | |
| 8. Subependymal giant cell astrocytoma | |
| 9. Cardiac rhabdomyoma | |
| 10. Lymphangioleiomyomatosis | |
| 11. Angiomyolipomas (≥ 2) |

")
There are no specific symptoms for the diagnosis of TSC. However, there are clinical presentations that can occur in the context of TSC that require further investigation. Moreover, if the patient under investigation has one of the biological parents with TSC, there is a 50% chance that the patient under investigation has the condition. This 50% risk remains unchanged when multiple siblings are affected.
Diagnostic suspicion of TSC comes from the antenatal detection of cardiac rhabdomyomas; the post-natal identification of hypopigmented macules on the skin; seizures in childhood, particularly with spasms; and presence or absence of cognitive impairment during autism assessment. The International Tuberous Sclerosis Complex Consensus recommends yearly screening for neuropsychiatric disorders and when clinically indicated.19
From the dermatologist point of view, a full dermatological examination is recommended. Family history of at least three generations should be obtained, and genetic testing should be considered in cases where there is suspicion of the diagnosis or for family-planning. Dental and ophthalmological evaluations are also recommended, including fundoscopy, to all individuals with the diagnosis of TSC in search of hamartomas and retinal hypopigmented lesions. Complementary investigations should be considered in children at the time of diagnosis and include brain magnetic resonance, to evaluate tubercles, subependymal nodules or other intracranial lesions; electroencephalogram, to evaluate the activity of subclinical seizure; transthoracic echocardiogram, to investigate rhabdomyomas (particularly in patients younger than three years of age) and electrocardiogram, to evaluate underlying conduction defects. Imaging studies, including abdominal ultrasound, computed tomography or magnetic resonance should be performed at the time of diagnosis, regardless of age, to evaluate angiomyolipomas or renal cysts, being the abdominal magnetic resonance the preferred method since many angiomyolipomas can be poor in fat tissue and not visualized by the other methods. Pulmonary function tests and high-resolution chest computed tomography for the investigation of lymphangioleiomyomatosis are indicated for women with 18 years old or older and in symptomatic men.21
TreatmentTSC treatment consists, above all, of the management of the symptoms caused by hamartomas and prophylactic measures to avoid loss of function of the affected organ. Because it is a systemic disease, a multidisciplinary follow-up is mandatory, with the need of assessment and follow-up in conjunction with teams of genetics, neurology, ophthalmology, pneumology, nephrology, and odontology.1
From the dermatological point of view, multiple descriptive or surgical treatments were developed to decrease the development and remove facial angiofibromas such as dermabrasion, surgical excision, electrocautery and laser. However, these procedures tend to be uncomfortable for the patient, need to be repeated periodically to avoid recurrence of the lesions and many times need to be associated to other therapeutic methods in an attempt to optimize results.1,13
For the treatment of facial angiofibromas with a predominantly vascular component, intense pulsed light (IPL) has been shown particularly effective. Fibrous or protruding lesions respond better to carbon dioxide laser resurfacing, although this treatment also presents a higher risk for hypertrophic scarring.22
Due to the progressive enlargement and recurrence of facial tumors, surgical treatment can be postponed until after adolescence, when their growth is maximum. Painful ungual fibromas can be surgically excised, cauterized or treated with laser, however, recurrence is common.1
In TSC patients, the mTOR protein is aberrantly activated in fibroblast -like cells located in the dermis. These cells produce an epidermal growth factor, epiregulin, that simulates cellular proliferation.13,23 This overproduction of cells along with angiogenesis result in the initial appearance and continuous progression of facial angiofibromas throughout life.
After the discovery of the regulation of the mTOR pathway in the development of TSC tumors and with the advent of the target therapy using mTORC1 inhibitors, some promising studies have been highlighted, favoring the possibility of treating TSC patients according to the physiopathogenesis of the condition. Rapamycin is a natural macrolide isolated from Streptomyces hygroscopicus in 1965, that binds specifically to mTOR, resulting in the inhibition of mTOR activity and finally promoting the inhibition of cellular growth.13 Rapamycin mTOR inhibitors and their derivative everolimus have been studied in TSC patients since 2006 and are promising for the treatment of multiple tumors including renal angiomyolipomas, giant cell subependymal astrocytomas and lymphangioleiomyomatosis, with secondary benefits on the cutaneous manifestations.24-26
Randomized, double-blind, placebo-controlled clinical trials, EXIST-1 (efficacy and safety of everolimus for subependymal giant cell astrocytomas – SEGAs - associated with tuberous sclerosis complex) and EXIST-2 (everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis) demonstrated beneficial effects of everolimus in both TSC manifestations.27,28 These studies allowed the approval of everolimus by the European Medicines Agency (EMA) and by the Food and Drug Administration (FDA) for the treatment of adult patients with renal angiomyolipoma associated to TSC and with risk of complications (based on the size of the tumor, the presence of aneurysm and of multiple or bilateral tumors) that did not need immediate surgery. Everolimus is also indicated for the treatment of patients with SEGAs regardless of age, who require therapeutic intervention but are not eligible for surgery.29 Either rapamycin or everolimus proved to reduce the severity of epilepsy in TSC patients.25,30 In Brazil, these drugs are approved by the Agência Nacional de Vigilância Sanitária (ANVISA) in patients with TSC with the following indications/ everolimus for renal angiomyolipomas without indication of immediate surgery (in patients over 18 years) and SEGA; and sirolimus for lymphangioleiomyomatosis.31,32
In regards to the cutaneous effects of mTOR inhibitors, in a retrospective study that evaluated the response to sirolimus in 14 patients with lymphangioleiomyomatosis, they were also submitted to serial photographic documentation of the facial angiofibromas before, during and after the treatment, associated to microscopic and molecular studies of these lesions before and during the administration of sirolimus. The study revealed that cutaneous tumors not only improved after treatment with systemic sirolimus but also maintained the improvement for at least 64 months of treatment.31 In a similar way to other studies that also evaluated the secondary cutaneous response to systemic sirolimus indicated for renal tumors, facial angiofibromas responded more favorably to the treatment than ungual fibromas and shagreen patches. Therefore, it is possible that the antiangiogenic properties of sirolimus are responsible for the apparent superior benefit in highly vascularized cutaneous tumors. Unexpectedly, angiofibromas did not worsen after discontinuation of the treatment in this study. This is contrary to the progression of cutaneous and visceral tumors reported in previous studies after ceasing systemic therapy.31,32
Current guidelines limit the use of oral mTORC1 inhibitors for the treatment of TSC cutaneous lesions for individuals that are not eligible to surgical approaches and whose skin lesions have a severe risk of recurrent and extensive hemorrhages.5
However, the oral treatment with mTOR inhibitors is associated to an increased risk of infections, stomatitis being the most frequent, acne, amenorrhea and laboratory abnormalities. Considerations of these effects are important, since treatment with mTOR inhibitors should be long lasting because withdrawal of the medication results in a rebound growth of the tumor (SEGA, renal angiomyolipoma, cardiac rhabdomyoma and cutaneous lesions) in the majority of the patients.33
A small series of cases reported benefits using topical preparations of rapamycin in facial angiofibromas, leading to the reduction in size and in the erythema and, in some cases, to the complete resolution of the cutaneous lesions. However, the long-term safety evidence is still scarce.5 Early mTOR inhibition can prevent the development of facial angiofibromas, as suggested by a case report in a female monozygotic twin treated with systemic everolimus for SEGA since she was four years of age. Her twin sister who was not treated developed facial angiofibromas when she was six years of age, while the patient treated did not.34
At last, it is known that many dermatological diseases cause a negative impact in the life of patients, due to stigmas caused by the appearance of cutaneous lesions. This not only affects their emotional state, but also their social relations and daily activities. In TSC patients, we observe this impact, so it is fundamental that the medical team that follows these patients to consider not only the clinical aspects of the disease, but also the psychologic and social morbidities inherent to this condition.35,36
Financial support: None.
Conflict of interests: None.
Questions1. Tuberous sclerosis complex is a genodermatosis:
A) Autosomal dominant with hamartomas that affect multiple organs.
B) Autosomal recessive with hamartomas that affect multiple organs.
C) Autosomal dominant predominantly affecting females, and the most commonly affected organs are skin, central nervous system, heart, lungs and kidneys.
D) Autosomal recessive with vascular malformations in many organs, being the most affected skin and central nervous system.
2. The classical tuberous sclerosis triad is characterized by:
A) Shagreen patch, facial angiofibromas and mental retardation.
B) Epilepsy, sebaceous adenoma and shagreen patch.
C) Facial angiofibromas, fibrous cephalic plaque and epilepsy.
D) Mental retardation, epilepsy and sebaceous adenoma (angiofibroma).
3. Choose the sentence with the correct sequence:
( ) Skin lesions are found in more than 90% of cases, the majority appearing in the 3rd decade of life.
( ) “Ash-leaf” lesions tend to follow Blaschko lines when on the trunk, typical for type 3 mosaicism.
( ) Hypopigmented macules in “confetti” or guttate leukoderma are hypopigmented macules that appear over the years.
( ) Hypopigmented macules are many times found since birth, allowing for the early diagnosis of the disease when associated to other clinical findings.
A) F T T F
B) F F F T
C) T T F T
D) F T F T
4. About connective tissue nevi and ungual fibromas or Koenen tumors, it is correct to say that:
A) Connective tissue nevi seen in TSC are normal-colored to brown plaques restricted to the lumbosacral region.
B) Shagreen patch many times have the aspect of orange peel and are found on the upper back.
C) Fibrous cephalic plaque is present in more than 80% of cases, being most commonly found on one side of the forehead.
D) Koenen tumor usually appears during adolescence and has a predilection for females and the toes.
5. According to the systemic manifestations of TSC, we can affirm that the correct items are:
1- Angiomyolipomas increase progressively from infancy and stop growing in the elderly.
2- Renal lesions have an indolent, clinically asymptomatic course, with no potentially severe repercussions in the patient’s life.
3- In cases of patients that evolve with hematuria in adulthood, the possibility of malignant transformation of the angiomyolipomas must be kept in mind, since they are usually asymptomatic.
4- Lymphangioleiomyomatosis affects predominantly younger women with the first manifestations after early adolescence.
5- Rhabdomyoma is the most common cardiac tumor and tends to grow progressively in the first year, when it stabilizes and continues until adulthood.
A) 1 - 3
B) 2 - 4
C) 3 - 5
D) 1 - 4
6. Choose the INCORRECT option:
A) The identification of the pathogenic TSC1 or TSC2 DNA mutation is not enough to establish the diagnosis of TSC.
B) Molecular testing yields a positive result in 75% to 90% of TSC patients.
C) In 10% to 25% of patients, conventional genetic testing does not identify the pathogenic TSC1 or TSC2 mutation.
D) The identification of the pathogenic mutation for TSC in conventional genetic testing configures an independent diagnostic criterion.
7. About the diagnostic criteria for the tuberous sclerosis complex, reviewed in 2012, in the second International Tuberous Sclerosis Complex Consensus Conference, all are included as extracutaneous manifestations, EXCEPT:
A) Subependymal giant cell astrocytoma.
B) Cardiac rhabdomyoma.
C) Lymphangioleiomyomatosis.
D) Pigmented hamartomas of the iris.
8. It is a MAJOR clinical criterion for the diagnosis of TSC:
A) ≥ 5 hypopigmented macules with at least 3mm diameter.
B) ≥ 3 facial angiofibromas.
C) ≥ 2 intraoral fibromas.
D) > 3 dental enamel pits.
9. From the dermatological point of view, many destructive or surgical treatment options are used to reduce the development of and remove facial angiofibromas. It is correct to say that:
A) The destructive procedures are usually comfortable for the patient and can be performed at any age.
B) It usually is not necessary to repeat laser treatments periodically because the lesions tend not to recur.
C) Are therapeutic options for facial angiofibromas dermabrasion, surgical excision, electrocautery and laser.
D) It is not necessary to combine therapeutic methods to optimize results since that regardless of the treatment chosen, all provide satisfactory results.
10. Diagnostic suspicion of TSC comes from the following findings, EXCEPT:
A) Antenatal detection of cardiac rhabdomyomas.
B) Post-natal identification of hypopigmented patches on the skin.
C) Visualization of ephelis on the axillary or inguinal regions.
D) Seizures in childhood, particularly with spasms.
Answer key
Gnathostomiasis: an emerging infectious disease relevant to all dermatologists. An Bras Dermatol. 2018;93(2): 172-80.
1. D
2. C
3. A
4. A
5. D
6. C
7. D
8. B
9. D
10. D
Papers
Information for all members: The EMC-D questionnaire is now available at the homepage of the Brazilian Annals of Dermatology: www.anaisdedermatologia.org.br. The deadline for completing the questionnaire is 30 days from the date of online publication.
