

Facomatose pigmento vascular (PPV) é anomalia congênita rara caracterizada pela presença de lesões vasculares e pigmentares. Apresentamos um caso de rara associação de cútis marmorata telangectásica congênita, nevus flammeus e manchas mongólicas aberrantes. Esses achados não permitem o enquadramento em nenhuma classificação de PPV, exceto na denominação proposta de facomatose cesio‐flammea‐marmorata.1
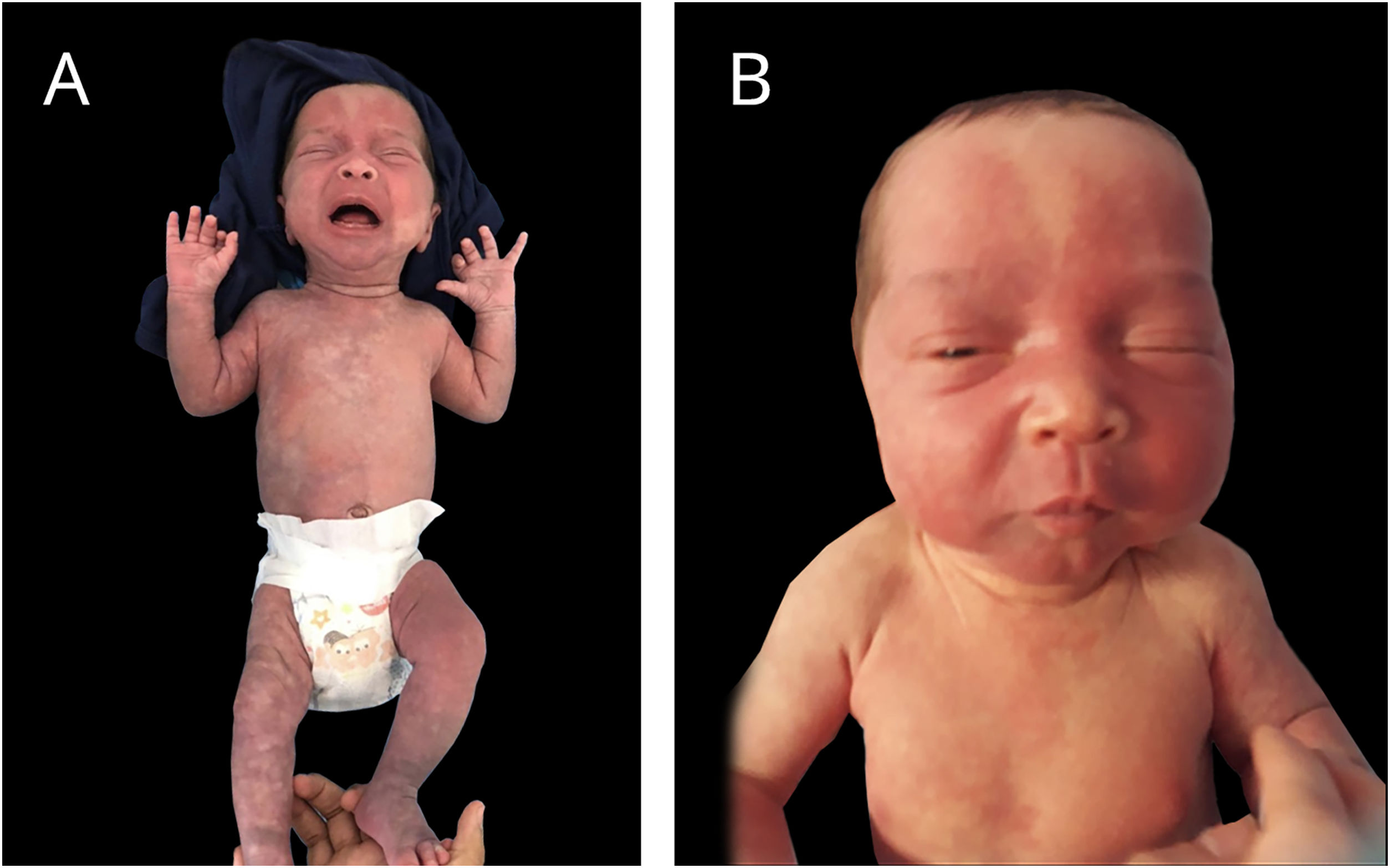
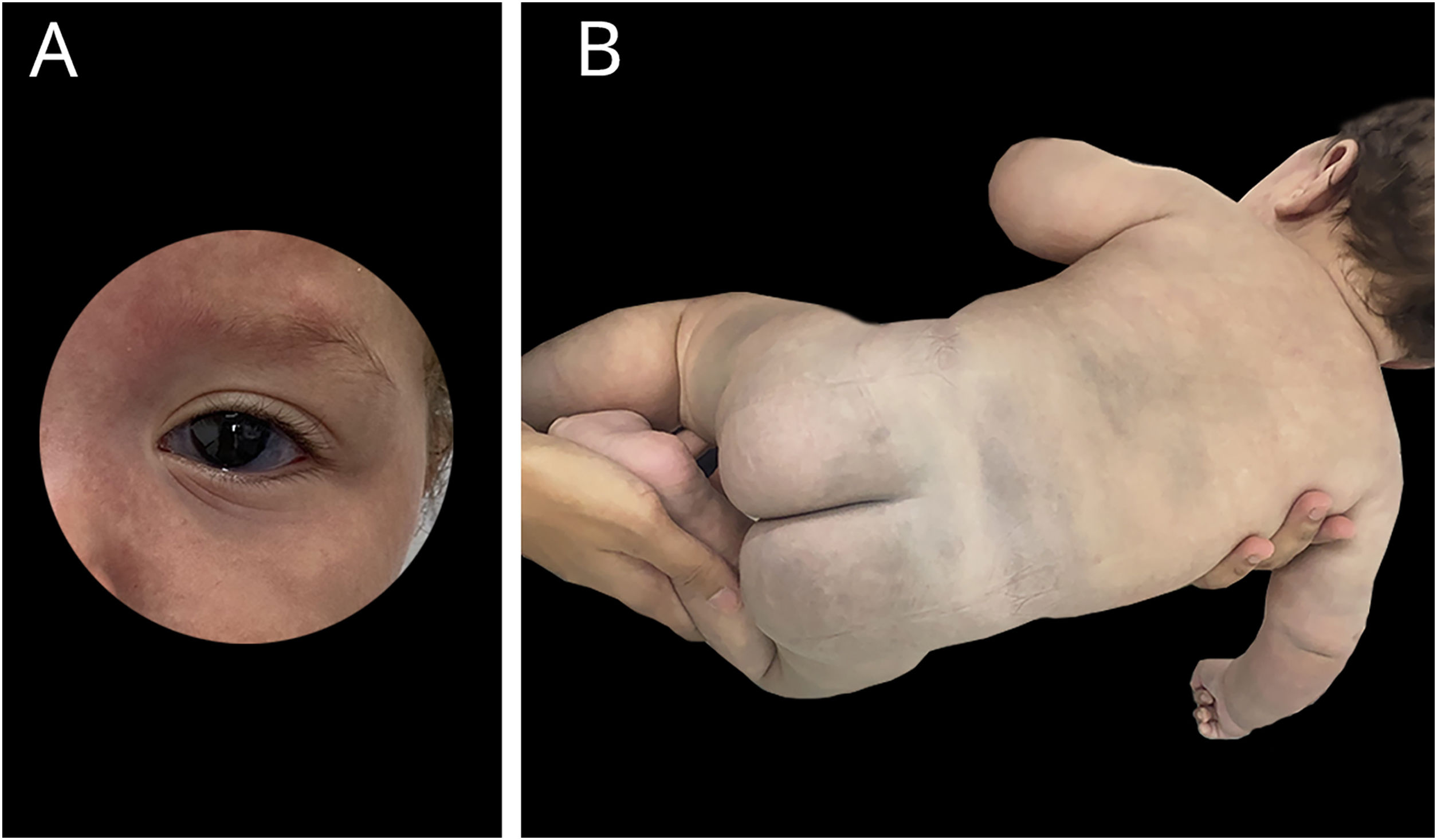
Bebê nascido pré‐termo tardio, de gestação gemelar dicoriônica e diamniótica, pais não consanguíneos, mãe teve quadro SARS‐CoV‐2 durante gestação; parto e gestação sem intercorrência. Irmão gêmeo sem alterações. Apresentava desde o nascimento lesão eritematosa acometendo quase a totalidade da face, compatível com nevus flammeus, eritema em padrão reticular em hemicorpo à direita com nítida demarcação em linha média anterior, compatível com cútis telangectásica marmorata congênita, extensas máculas azul‐acinzentadas nos membros inferiores, glúteos e dorso, compatíveis com manchas mongólicas. Apresentava, ainda, escleras acinzentadas bilateralmente e discreta assimetria na circunferência de membros inferiores. As lesões vasculares se tornavam mais intensas ao choro ou exposição ao frio (figs. 1 e 2).


Realizados ecocardiograma, ultrassonografia transfontanela e abdominal, sem alterações. Avaliação neurológica, oftálmica e ortopédica, sem anormalidades. Ultrassom dermatológico identificou hipoplasia de tecido subcutâneo na perna direita em comparação ao membro contralateral.
O sequenciamento completo de exoma em sangue periférico identificou variante provavelmente patogênica em heterozigose no gene COL17A1, (c.3277+1G>A), além de variante patogênica em heterozigose no gene HBB, (c.20A>T: p., Glu7Val) atribuindo betalassemia minor.
No momento desta publicação, o paciente estava com 1 ano de idade e apresentava desenvolvimento neuropsicomotor normal e melhora parcial das lesões vasculares. Foi feito o diagnóstico de facomatose pigmento vascular; todavia, os achados não se alinham completamente aos critérios vigentes, o que tornou a classificação desafiadora. A denominação de facomatose cesio‐flammea‐marmorata já foi proposta na literatura, com a identificação de sete casos até o momento,1 que apresentam heterogeneidade de características, e um caso com a tríade descrita associada a nevo de Ota e nevo anêmico.2
FacomatosePPV é um grupo de síndromes congênitas raras caracterizadas pela combinação de malformações capilares e lesões pigmentadas, associadas ou não a manifestações sistêmicas. Inicialmente foi classificada em quatro tipos conforme os fenótipos (I ‐ Nevus flammeus e nevus pigmentosos ou verrucoso; II ‐ flammeus +/− anêmico e manchas mongólicas; III ‐ flammeus +/− anêmico e spilus; IV ‐ flammeus +/− anêmico e spilus com mancha mongólica) e em subtipos (a – acometimento apenas cutâneo; b – acometimento sistêmico). Happle3 reclassificou em facomatose cesioflamea, cesiomarmorata, spilorosea e sem classificação, com ou sem manifestação sistêmica.
A patogênese da síndrome não é bem compreendida. Os genes GNAG e GNA11 foram associados a alguns casos.4 O gene COL17A1, identificado neste paciente, codifica o colágeno XVII, essencial na estabilização da epiderme e, até onde sabemos, é a primeira vez que variantes possivelmente patogênicas neste gene são identificadas em um paciente com essas características. Esse gene é relacionado à epidermólise bolhosa; contudo, não tem penetrância de 100%, o que pode explicar a ausência de fenótipo. O paciente não apresenta fragilidade cutânea até o momento.
O diagnóstico de PPV é clínico; entretanto, a classificação é desafiadora quando não preenche todos os critérios ou há uma sobreposição entre eles, motivo pelo qual já foi proposto que a combinação das três condições encontradas neste paciente receba uma classificação distinta como facomatose cesio‐flammeo‐marmorata.1 Recomenda‐se investigação de acometimento ocular e neurológico,5 principalmente, e outros exames, conforme suspeição clínica.
Suporte financeiroNenhum.
Contribuição dos autoresLuciane Zagonel: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura.
Mônica Vannucci Nunes Lipay: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura.
Glaucos Ricardo Paraluppi: Aprovação da versão final do manuscrito; revisão crítica do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica do caso estudado.
Célia Antônia Xavier de Moraes Alves: Aprovação da versão final do manuscrito; revisão crítica do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica do caso estudado.
Conflito de interessesNenhum.
AgradecimentoDra. Rosa Sigrist.
Como citar este artigo: Zagonel L, Lipay MVN, Paraluppi GR, Alves CAXM. Phacomatosis cesio‐flammeo‐marmorata: report of a rare association. An Bras Dermatol. 2024;99:462–4.
Trabalho realizado na Faculdade de Medicina de Jundiaí, Jundiaí, SP, Brasil.
