






The term vasculitis refers to the inflammation of vessel walls. It may range in severity from a self-limited disorder in one single organ to a life-threatening disease due to multiple organ failure. It has many causes, although they result in only a few histological patterns of vascular inflammation. Vessels of any type and in any organ can be affected, a fact that results in a broad variety of signs and symptoms. Different vasculitides with indistinguishable clinical presentations have quite different prognosis and treatments. This condition presents many challenges to physicians in terms of classification, diagnosis, appropriate laboratory workup, and treatment. Moreover, it compels a careful follow-up. This article reviews the Chapel-Hill 2012 classification, etiology, recent insights in pathophysiology, some important dermatological clues for the diagnosis and summarizes treatment of some of these complex vasculitis syndromes.
Vasculitides encompass a large group of heterogeneous diseases characterized by inflammatory reaction localized in the vascular wall and perivascular tissues. To the concern of etiology, it is defined as a complex syndrome because several types of vasculitides can be elicited by different stimulus, although most of them are classified as idiopathic. Both, venous or arterial vessels can be affected, in a pattern of single organ or multisystemic disease. Then, dealing with vasculitides often means to study several distinct pathophysiological mechanisms and a wide clinical spectrum. Often several authors rely on the blood vessel caliber to classify them as small, medium or large-vessel vasculitis. However, when it is held alone, this classification fails to recognize diversity within vessels of the same caliber, their specialized roles in several anatomic areas in response to stimuli, injury, and repair that determine disease patterns.1,2
Cutaneous vasculitides can range in severity from benign, self-limited, short-course skin eruption to life-threatening disease with multiple-organ failure. Otherwise, systemic vasculitis is divided into two main categories: primary vasculitis syndrome and secondary vasculitis syndrome. The first one is caused by unknown etiology inflammation of blood vessels, while the second one is induced by underlying conditions, including connective tissue diseases, cancer, infections, immunization, and drug allergy. In the majority of cases the cutaneous vasculitides syndromes will show a neutrophilic small vessel vasculitis, often called Cutaneous Leukocytoclastic Angiitis (CLA).3
The aim of this review is to clarify the recent concepts about the etiology of distinct vasculitides, the classification preconized for the Chapel Hill Consensus Conference (CHCC) and to emphasize the clues on dermatological physical and/or histopathological exam that may help to suggest a specific group of vasculitis in the clinical practice.
Classification of vasculitisClinicians have found it convenient to use vessel size as the most important feature to distinguish different forms of vasculitides.2 The CHCC published in 2012 proposed the unification of new types of vasculitis syndromes and their associated illness (Table 1).4 However, CHCC is only a nomenclature system/nosology and not a classification or a diagnostic system that helps to direct clinical management. Thus, it subdivides vasculitides based on combinations of features that separate different conditions into definable categories.4
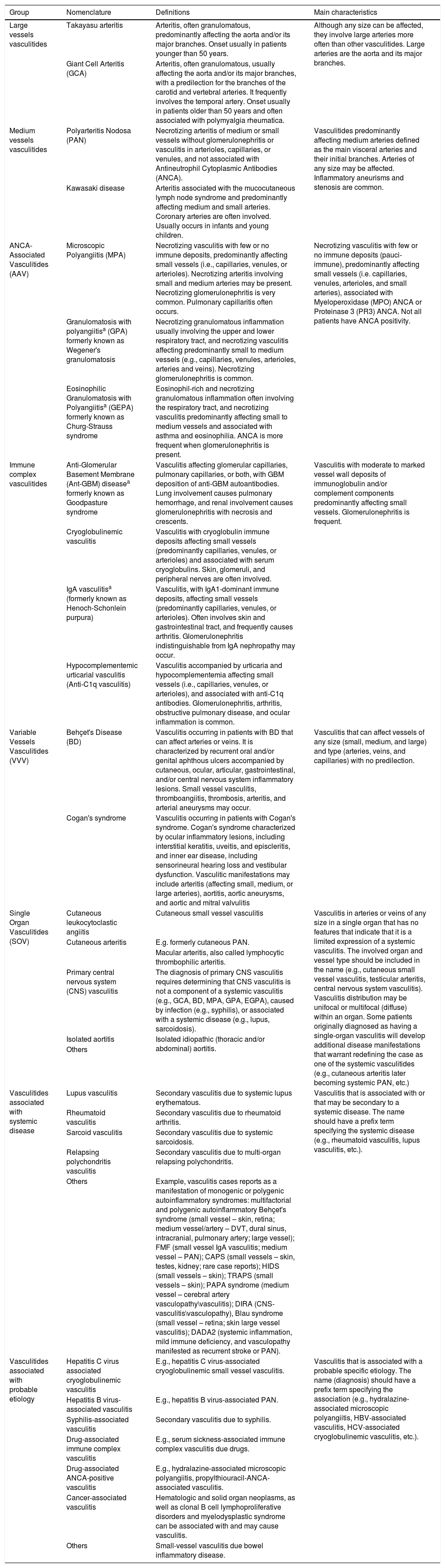
Classification and definitions of vasculitides adapted from data adopted by the 2012 Chapel Hill Consensus Conference.
| Group | Nomenclature | Definitions | Main characteristics |
|---|---|---|---|
| Large vessels vasculitides | Takayasu arteritis | Arteritis, often granulomatous, predominantly affecting the aorta and/or its major branches. Onset usually in patients younger than 50 years. | Although any size can be affected, they involve large arteries more often than other vasculitides. Large arteries are the aorta and its major branches. |
| Giant Cell Arteritis (GCA) | Arteritis, often granulomatous, usually affecting the aorta and/or its major branches, with a predilection for the branches of the carotid and vertebral arteries. It frequently involves the temporal artery. Onset usually in patients older than 50 years and often associated with polymyalgia rheumatica. | ||
| Medium vessels vasculitides | Polyarteritis Nodosa (PAN) | Necrotizing arteritis of medium or small vessels without glomerulonephritis or vasculitis in arterioles, capillaries, or venules, and not associated with Antineutrophil Cytoplasmic Antibodies (ANCA). | Vasculitides predominantly affecting medium arteries defined as the main visceral arteries and their initial branches. Arteries of any size may be affected. Inflammatory aneurisms and stenosis are common. |
| Kawasaki disease | Arteritis associated with the mucocutaneous lymph node syndrome and predominantly affecting medium and small arteries. Coronary arteries are often involved. Usually occurs in infants and young children. | ||
| ANCA-Associated Vasculitides (AAV) | Microscopic Polyangiitis (MPA) | Necrotizing vasculitis with few or no immune deposits, predominantly affecting small vessels (i.e., capillaries, venules, or arterioles). Necrotizing arteritis involving small and medium arteries may be present. Necrotizing glomerulonephritis is very common. Pulmonary capillaritis often occurs. | Necrotizing vasculitis with few or no immune deposits (pauci-immune), predominantly affecting small vessels (i.e. capillaries, venules, arterioles, and small arteries), associated with Myeloperoxidase (MPO) ANCA or Proteinase 3 (PR3) ANCA. Not all patients have ANCA positivity. |
| Granulomatosis with polyangiitisa (GPA) formerly known as Wegener's granulomatosis | Necrotizing granulomatous inflammation usually involving the upper and lower respiratory tract, and necrotizing vasculitis affecting predominantly small to medium vessels (e.g., capillaries, venules, arterioles, arteries and veins). Necrotizing glomerulonephritis is common. | ||
| Eosinophilic Granulomatosis with Polyangiitisa (GEPA) formerly known as Churg-Strauss syndrome | Eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels and associated with asthma and eosinophilia. ANCA is more frequent when glomerulonephritis is present. | ||
| Immune complex vasculitides | Anti-Glomerular Basement Membrane (Ant-GBM) diseasea formerly known as Goodpasture syndrome | Vasculitis affecting glomerular capillaries, pulmonary capillaries, or both, with GBM deposition of anti-GBM autoantibodies. Lung involvement causes pulmonary hemorrhage, and renal involvement causes glomerulonephritis with necrosis and crescents. | Vasculitis with moderate to marked vessel wall deposits of immunoglobulin and/or complement components predominantly affecting small vessels. Glomerulonephritis is frequent. |
| Cryoglobulinemic vasculitis | Vasculitis with cryoglobulin immune deposits affecting small vessels (predominantly capillaries, venules, or arterioles) and associated with serum cryoglobulins. Skin, glomeruli, and peripheral nerves are often involved. | ||
| IgA vasculitisa (formerly known as Henoch-Schonlein purpura) | Vasculitis, with IgA1-dominant immune deposits, affecting small vessels (predominantly capillaries, venules, or arterioles). Often involves skin and gastrointestinal tract, and frequently causes arthritis. Glomerulonephritis indistinguishable from IgA nephropathy may occur. | ||
| Hypocomplementemic urticarial vasculitis (Anti-C1q vasculitis) | Vasculitis accompanied by urticaria and hypocomplementemia affecting small vessels (i.e., capillaries, venules, or arterioles), and associated with anti-C1q antibodies. Glomerulonephritis, arthritis, obstructive pulmonary disease, and ocular inflammation is common. | ||
| Variable Vessels Vasculitides (VVV) | Behçet's Disease (BD) | Vasculitis occurring in patients with BD that can affect arteries or veins. It is characterized by recurrent oral and/or genital aphthous ulcers accompanied by cutaneous, ocular, articular, gastrointestinal, and/or central nervous system inflammatory lesions. Small vessel vasculitis, thromboangiitis, thrombosis, arteritis, and arterial aneurysms may occur. | Vasculitis that can affect vessels of any size (small, medium, and large) and type (arteries, veins, and capillaries) with no predilection. |
| Cogan's syndrome | Vasculitis occurring in patients with Cogan's syndrome. Cogan's syndrome characterized by ocular inflammatory lesions, including interstitial keratitis, uveitis, and episcleritis, and inner ear disease, including sensorineural hearing loss and vestibular dysfunction. Vasculitic manifestations may include arteritis (affecting small, medium, or large arteries), aortitis, aortic aneurysms, and aortic and mitral valvulitis | ||
| Single Organ Vasculitides (SOV) | Cutaneous leukocytoclastic angiitis | Cutaneous small vessel vasculitis | Vasculitis in arteries or veins of any size in a single organ that has no features that indicate that it is a limited expression of a systemic vasculitis. The involved organ and vessel type should be included in the name (e.g., cutaneous small vessel vasculitis, testicular arteritis, central nervous system vasculitis). Vasculitis distribution may be unifocal or multifocal (diffuse) within an organ. Some patients originally diagnosed as having a single-organ vasculitis will develop additional disease manifestations that warrant redefining the case as one of the systemic vasculitides (e.g., cutaneous arteritis later becoming systemic PAN, etc.) |
| Cutaneous arteritis | E.g. formerly cutaneous PAN. | ||
| Macular arteritis, also called lymphocytic thrombophilic arteritis. | |||
| Primary central nervous system (CNS) vasculitis | The diagnosis of primary CNS vasculitis requires determining that CNS vasculitis is not a component of a systemic vasculitis (e.g., GCA, BD, MPA, GPA, EGPA), caused by infection (e.g., syphilis), or associated with a systemic disease (e.g., lupus, sarcoidosis). | ||
| Isolated aortitis | Isolated idiopathic (thoracic and/or abdominal) aortitis. | ||
| Others | |||
| Vasculitides associated with systemic disease | Lupus vasculitis | Secondary vasculitis due to systemic lupus erythematous. | Vasculitis that is associated with or that may be secondary to a systemic disease. The name should have a prefix term specifying the systemic disease (e.g., rheumatoid vasculitis, lupus vasculitis, etc.). |
| Rheumatoid vasculitis | Secondary vasculitis due to rheumatoid arthritis. | ||
| Sarcoid vasculitis | Secondary vasculitis due to systemic sarcoidosis. | ||
| Relapsing polychondritis vasculitis | Secondary vasculitis due to multi-organ relapsing polychondritis. | ||
| Others | Example, vasculitis cases reports as a manifestation of monogenic or polygenic autoinflammatory syndromes: multifactorial and polygenic autoinflammatory Behçet's syndrome (small vessel – skin, retina; medium vessel/artery – DVT, dural sinus, intracranial, pulmonary artery; large vessel); FMF (small vessel IgA vasculitis; medium vessel – PAN); CAPS (small vessels – skin, testes, kidney; rare case reports); HIDS (small vessels – skin); TRAPS (small vessels – skin); PAPA syndrome (medium vessel – cerebral artery vasculopathy\vasculitis); DIRA (CNS-vasculitis\vasculopathy), Blau syndrome (small vessel – retina; skin large vessel vasculitis); DADA2 (systemic inflammation, mild immune deficiency, and vasculopathy manifested as recurrent stroke or PAN). | ||
| Vasculitides associated with probable etiology | Hepatitis C virus associated cryoglobulinemic vasculitis | E.g., hepatitis C virus-associated cryoglobulinemic small vessel vasculitis. | Vasculitis that is associated with a probable specific etiology. The name (diagnosis) should have a prefix term specifying the association (e.g., hydralazine-associated microscopic polyangiitis, HBV-associated vasculitis, HCV-associated cryoglobulinemic vasculitis, etc.). |
| Hepatitis B virus-associated vasculitis | E.g., hepatitis B virus-associated PAN. | ||
| Syphilis-associated vasculitis | Secondary vasculitis due to syphilis. | ||
| Drug-associated immune complex vasculitis | E.g., serum sickness-associated immune complex vasculitis due drugs. | ||
| Drug-associated ANCA-positive vasculitis | E.g., hydralazine-associated microscopic polyangiitis, propylthiouracil-ANCA-associated vasculitis. | ||
| Cancer-associated vasculitis | Hematologic and solid organ neoplasms, as well as clonal B cell lymphoproliferative disorders and myelodysplastic syndrome can be associated with and may cause vasculitis. | ||
| Others | Small-vessel vasculitis due bowel inflammatory disease. |
New nomenclature: DADA2, Deficiency of Adenosine Deaminase type 2; DIRA, Deficiency of Interleukin-1 Receptor Antagonist; DVT, Deep Venous Thromboembolism; FMF, Familial Mediterranean Fever; HIDS, Hyper IgD with periodic fever Syndrome; PAPA, Pyogenic Arthritis, Pyoderma gangrenosum, and Acne; TRAPS, TNF Receptor-Associated Periodic Syndrome.
Some cutaneous manifestations involving small, medium and/or large vessels can be found not only in the autoimmune diseases, but also in the autoinflammatory diseases. This term was coined to describe an emerging family of conditions characterized by episodes of unprovoked inflammation due to dysregulation of the innate immune system without the primary role of autoreactive T lymphocytes and/or autoantibodies. Skin involvement in autoinflammatory diseases generally resembles urticarial vasculitis or may have features of neutrophilic dermatoses. Additional skin lesions include purpuric exanthema, nonthrombocytopenic purpura, and pyoderma gangrenosum. In some autoinflammatory diseases, it is still unclear whether vasculitis is an integrated clinical manifestation or represents an additional independent disease.5 We have also included these conditions in a proper sub-classification in table 1.
Generally, the size of vessel involvement correlates with clinical morphology on the dermatological exam. Small, predominately superficial vessel involvement results in urticaria, infiltrated erythema, palpable or non-palpable purpura, vesiculobullous and/or pustules lesions. Ulcers, nodules, pitted scars, white atrophy, or livedo racemosa are associated with arterial-muscular vessel involvement, which will be located at dermal-hypodermal interface or within the subcutis. In vasculitides that involve small and medium-size vessels all kind of cutaneous lesions previously described can co-exist in the same patient. Finally, a large number of ulcers, especially when they are extensive and associated with necrosis, which may be consequence of deep-vessel arterial involvement, often predict recurrent vasculitis and systemic disease (Fig. 1).6
, types of blood vessels found in upper and lower dermis vascular plexus, vessels diameter, site of main vasculitis syndromes and sufficient depth of biopsies to represent the skin layer in which the disease is localized.")
Schematic representation of the skin (epidermis, dermis and subcutaneous tissue), types of blood vessels found in upper and lower dermis vascular plexus, vessels diameter, site of main vasculitis syndromes and sufficient depth of biopsies to represent the skin layer in which the disease is localized.
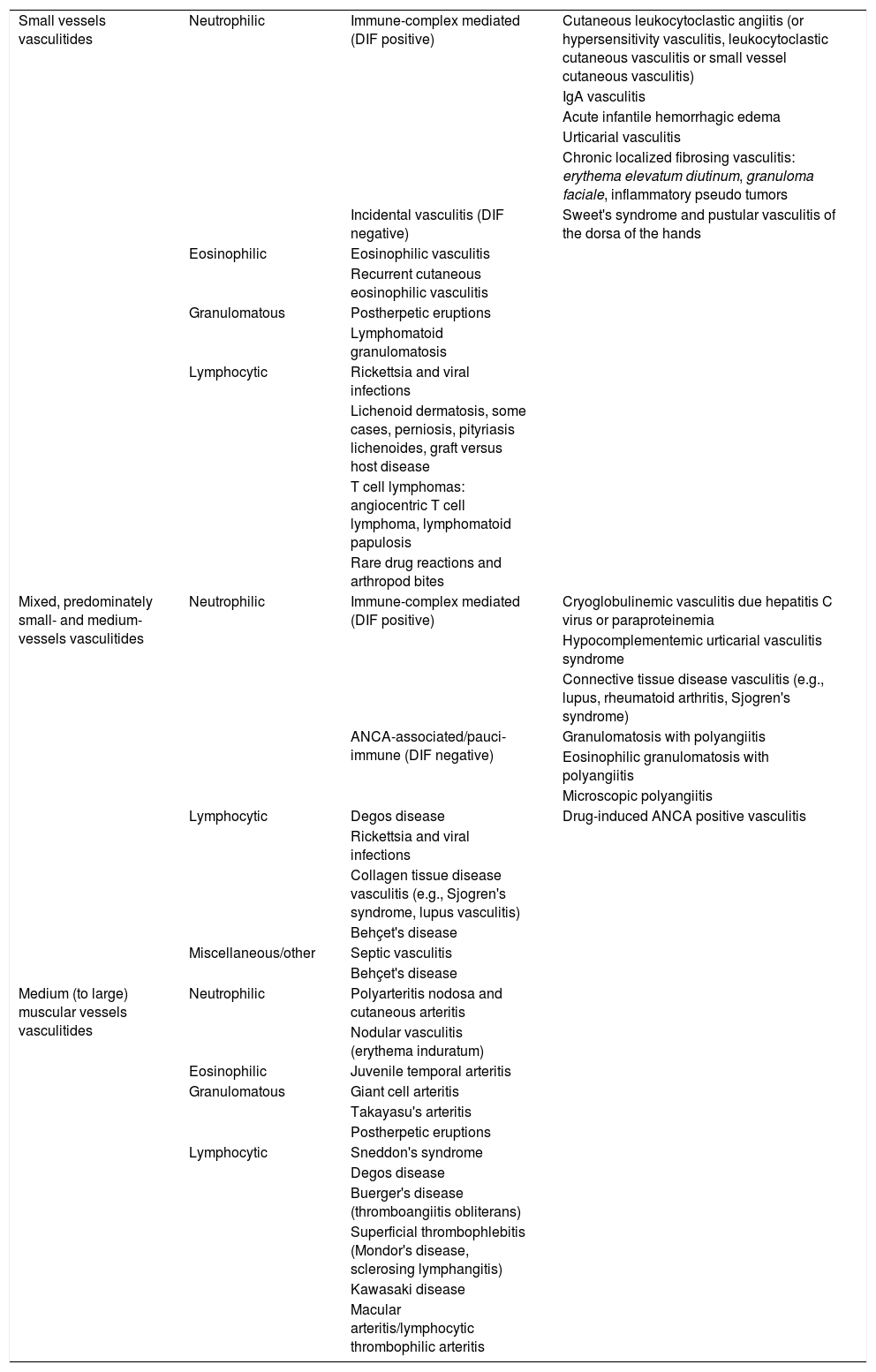
The recognition of pathologic changes in a cutaneous biopsy enables the identification distinct vasculitis conditions and signals the possibility of progression from localized cutaneous to systemic vasculitis.6 Skin biopsy is the gold standard method for the diagnosis of cutaneous vasculitis, also allowing differential diagnoses from vasculitis mimics, such as vaso-occlusive conditions and other diseases (Table 2).
Classification of vasculitides according to the vessel size and histopathological findings in biopsy specimens.
| Small vessels vasculitides | Neutrophilic | Immune-complex mediated (DIF positive) | Cutaneous leukocytoclastic angiitis (or hypersensitivity vasculitis, leukocytoclastic cutaneous vasculitis or small vessel cutaneous vasculitis) |
| IgA vasculitis | |||
| Acute infantile hemorrhagic edema | |||
| Urticarial vasculitis | |||
| Chronic localized fibrosing vasculitis: erythema elevatum diutinum, granuloma faciale, inflammatory pseudo tumors | |||
| Incidental vasculitis (DIF negative) | Sweet's syndrome and pustular vasculitis of the dorsa of the hands | ||
| Eosinophilic | Eosinophilic vasculitis | ||
| Recurrent cutaneous eosinophilic vasculitis | |||
| Granulomatous | Postherpetic eruptions | ||
| Lymphomatoid granulomatosis | |||
| Lymphocytic | Rickettsia and viral infections | ||
| Lichenoid dermatosis, some cases, perniosis, pityriasis lichenoides, graft versus host disease | |||
| T cell lymphomas: angiocentric T cell lymphoma, lymphomatoid papulosis | |||
| Rare drug reactions and arthropod bites | |||
| Mixed, predominately small- and medium-vessels vasculitides | Neutrophilic | Immune-complex mediated (DIF positive) | Cryoglobulinemic vasculitis due hepatitis C virus or paraproteinemia |
| Hypocomplementemic urticarial vasculitis syndrome | |||
| Connective tissue disease vasculitis (e.g., lupus, rheumatoid arthritis, Sjogren's syndrome) | |||
| ANCA-associated/pauci-immune (DIF negative) | Granulomatosis with polyangiitis | ||
| Eosinophilic granulomatosis with polyangiitis | |||
| Microscopic polyangiitis | |||
| Drug-induced ANCA positive vasculitis | |||
| Lymphocytic | Degos disease | ||
| Rickettsia and viral infections | |||
| Collagen tissue disease vasculitis (e.g., Sjogren's syndrome, lupus vasculitis) | |||
| Behçet's disease | |||
| Miscellaneous/other | Septic vasculitis | ||
| Behçet's disease | |||
| Medium (to large) muscular vessels vasculitides | Neutrophilic | Polyarteritis nodosa and cutaneous arteritis | |
| Nodular vasculitis (erythema induratum) | |||
| Eosinophilic | Juvenile temporal arteritis | ||
| Granulomatous | Giant cell arteritis | ||
| Takayasu's arteritis | |||
| Postherpetic eruptions | |||
| Lymphocytic | Sneddon's syndrome | ||
| Degos disease | |||
| Buerger's disease (thromboangiitis obliterans) | |||
| Superficial thrombophlebitis (Mondor's disease, sclerosing lymphangitis) | |||
| Kawasaki disease | |||
| Macular arteritis/lymphocytic thrombophilic arteritis |
ANCA, antineutrophil cytoplasmatic antibody; DIF positive, direct immunofluorescence examination of the skin lesions showing vessel wall immune-complexes and/or complement deposition.
The clinicians need to keep in mind that cutaneous vasculitis, particularly Cutaneous Small Vessel Vasculitis (CSVV), is often the most common presentation of several vasculitis syndromes.1 It may occur in 3% of Granulomatosis with Polyangiitis (GPA), 13% of Microscopic Polyangiitis (MPA), and 28% of Eosinophilic Granulomatosis with Polyangiitis (EGPA) patients.7 Some steps are essential in the clinical practice for accurate assistance to the patients with vasculitis: (i) meticulous anamnesis and physical exam; (ii) inspection and palpation of the cutaneous and subcutaneous lesions; (iii) a proper cutaneous biopsy is necessary according to the findings of dermatological exam; (iv) accurate propaedeutic and complementary exams must be performed to exclude systemic or internal organ involvement; (v) a work-up including indicated laboratory exams for infectious and parasitic infestations, autoimmune collagen diseases, blood marrow neoplasia and/or organ-solid cancer is useful for the evaluation of its etiology.
An important definition about the nosology of vasculitis in the skin was revised by Sunderkötter et al.1 The authors emphasized that they can be found in distinct clinical settings: (i) a cutaneous component of systemic vasculitides (e.g. cutaneous manifestations of IgA vasculitis), (ii) a skin-limited or skin-dominant expression or variant of a systemic vasculitis (e.g. skin-limited IgA vasculitis), or (iii) a Single-Organ Cutaneous Vasculitis (SOCV) that differs with regard to clinical, laboratory, and pathologic features from recognized systemic vasculitis (e.g. nodular vasculitis). SOCV does not develop full systemic vasculitis, while skin-dominant forms may do it. Then, systemic vasculitides are defined as those one presenting in at least one organ in addition to the skin.
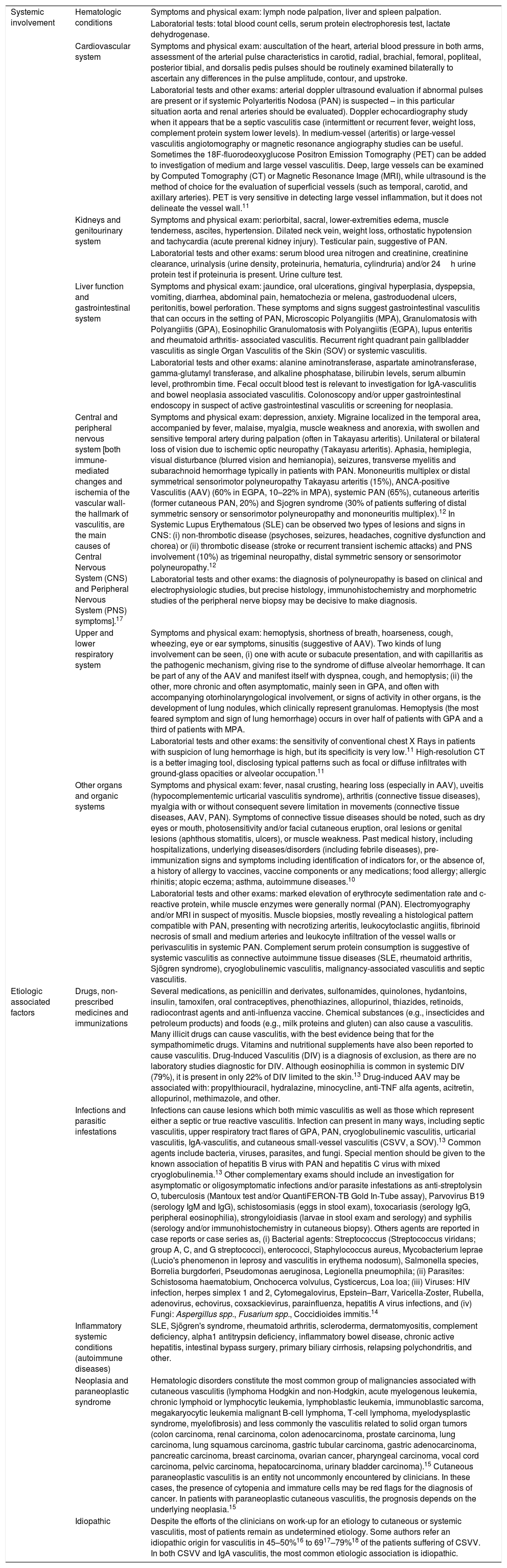
In most cases of CSVV, although nonspecific signs or symptoms of systemic inflammatory reactions, such as leukocytosis, raised C-reactive protein, or arthralgia may occur, significant systemic manifestations are unlikely.1 After a thorough history, review of systems and physical examination, a systematic and targeted laboratory workup should proceed to confirm SOCV. Although no standard protocol for this workup exists, screening tests should aim at elucidating the underlying cause and extent of organ involvement and should be guided by clinical signs and symptoms. Special attention should be paid to any evidence of systemic vasculitis, such as fever, weight loss, and other constitutional symptoms; arthritis; myalgias; abdominal pain, melena, or hematochezia; cough, hemoptysis, or dyspnea; hematuria or frothy urine; sinusitis or rhinitis; and paresthesia, weakness, or foot drop. Clinicians should also ask patients about potential triggers, including preceding infectious symptoms, usage of medications, vaccines and comorbidities.8–10 We summarized in table 3 the red flag symptoms and/or signs, complementary laboratory exams and etiologic factors to workup systemic vasculitis.11,12
Symptoms and signs evaluated to establish the diagnosis of systemic vasculitis and complementary exams needed to identify etiologic factors associated.
| Systemic involvement | Hematologic conditions | Symptoms and physical exam: lymph node palpation, liver and spleen palpation. |
| Laboratorial tests: total blood count cells, serum protein electrophoresis test, lactate dehydrogenase. | ||
| Cardiovascular system | Symptoms and physical exam: auscultation of the heart, arterial blood pressure in both arms, assessment of the arterial pulse characteristics in carotid, radial, brachial, femoral, popliteal, posterior tibial, and dorsalis pedis pulses should be routinely examined bilaterally to ascertain any differences in the pulse amplitude, contour, and upstroke. | |
| Laboratorial tests and other exams: arterial doppler ultrasound evaluation if abnormal pulses are present or if systemic Polyarteritis Nodosa (PAN) is suspected – in this particular situation aorta and renal arteries should be evaluated). Doppler echocardiography study when it appears that be a septic vasculitis case (intermittent or recurrent fever, weight loss, complement protein system lower levels). In medium-vessel (arteritis) or large-vessel vasculitis angiotomography or magnetic resonance angiography studies can be useful. Sometimes the 18F-fluorodeoxyglucose Positron Emission Tomography (PET) can be added to investigation of medium and large vessel vasculitis. Deep, large vessels can be examined by Computed Tomography (CT) or Magnetic Resonance Image (MRI), while ultrasound is the method of choice for the evaluation of superficial vessels (such as temporal, carotid, and axillary arteries). PET is very sensitive in detecting large vessel inflammation, but it does not delineate the vessel wall.11 | ||
| Kidneys and genitourinary system | Symptoms and physical exam: periorbital, sacral, lower-extremities edema, muscle tenderness, ascites, hypertension. Dilated neck vein, weight loss, orthostatic hypotension and tachycardia (acute prerenal kidney injury). Testicular pain, suggestive of PAN. | |
| Laboratorial tests and other exams: serum blood urea nitrogen and creatinine, creatinine clearance, urinalysis (urine density, proteinuria, hematuria, cylindruria) and/or 24h urine protein test if proteinuria is present. Urine culture test. | ||
| Liver function and gastrointestinal system | Symptoms and physical exam: jaundice, oral ulcerations, gingival hyperplasia, dyspepsia, vomiting, diarrhea, abdominal pain, hematochezia or melena, gastroduodenal ulcers, peritonitis, bowel perforation. These symptoms and signs suggest gastrointestinal vasculitis that can occurs in the setting of PAN, Microscopic Polyangiitis (MPA), Granulomatosis with Polyangiitis (GPA), Eosinophilic Granulomatosis with Polyangiitis (EGPA), lupus enteritis and rheumatoid arthritis- associated vasculitis. Recurrent right quadrant pain gallbladder vasculitis as single Organ Vasculitis of the Skin (SOV) or systemic vasculitis. | |
| Laboratorial tests and other exams: alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, and alkaline phosphatase, bilirubin levels, serum albumin level, prothrombin time. Fecal occult blood test is relevant to investigation for IgA-vasculitis and bowel neoplasia associated vasculitis. Colonoscopy and/or upper gastrointestinal endoscopy in suspect of active gastrointestinal vasculitis or screening for neoplasia. | ||
| Central and peripheral nervous system [both immune-mediated changes and ischemia of the vascular wall- the hallmark of vasculitis, are the main causes of Central Nervous System (CNS) and Peripheral Nervous System (PNS) symptoms].17 | Symptoms and physical exam: depression, anxiety. Migraine localized in the temporal area, accompanied by fever, malaise, myalgia, muscle weakness and anorexia, with swollen and sensitive temporal artery during palpation (often in Takayasu arteritis). Unilateral or bilateral loss of vision due to ischemic optic neuropathy (Takayasu arteritis). Aphasia, hemiplegia, visual disturbance (blurred vision and hemianopia), seizures, transverse myelitis and subarachnoid hemorrhage typically in patients with PAN. Mononeuritis multiplex or distal symmetrical sensorimotor polyneuropathy Takayasu arteritis (15%), ANCA-positive Vasculitis (AAV) (60% in EGPA, 10–22% in MPA), systemic PAN (65%), cutaneous arteritis (former cutaneous PAN, 20%) and Sjogren syndrome (30% of patients suffering of distal symmetric sensory or sensorimotor polyneuropathy and mononeuritis multiplex).12 In Systemic Lupus Erythematous (SLE) can be observed two types of lesions and signs in CNS: (i) non-thrombotic disease (psychoses, seizures, headaches, cognitive dysfunction and chorea) or (ii) thrombotic disease (stroke or recurrent transient ischemic attacks) and PNS involvement (10%) as trigeminal neuropathy, distal symmetric sensory or sensorimotor polyneuropathy.12 | |
| Laboratorial tests and other exams: the diagnosis of polyneuropathy is based on clinical and electrophysiologic studies, but precise histology, immunohistochemistry and morphometric studies of the peripheral nerve biopsy may be decisive to make diagnosis. | ||
| Upper and lower respiratory system | Symptoms and physical exam: hemoptysis, shortness of breath, hoarseness, cough, wheezing, eye or ear symptoms, sinusitis (suggestive of AAV). Two kinds of lung involvement can be seen, (i) one with acute or subacute presentation, and with capillaritis as the pathogenic mechanism, giving rise to the syndrome of diffuse alveolar hemorrhage. It can be part of any of the AAV and manifest itself with dyspnea, cough, and hemoptysis; (ii) the other, more chronic and often asymptomatic, mainly seen in GPA, and often with accompanying otorhinolaryngological involvement, or signs of activity in other organs, is the development of lung nodules, which clinically represent granulomas. Hemoptysis (the most feared symptom and sign of lung hemorrhage) occurs in over half of patients with GPA and a third of patients with MPA. | |
| Laboratorial tests and other exams: the sensitivity of conventional chest X Rays in patients with suspicion of lung hemorrhage is high, but its specificity is very low.11 High-resolution CT is a better imaging tool, disclosing typical patterns such as focal or diffuse infiltrates with ground-glass opacities or alveolar occupation.11 | ||
| Other organs and organic systems | Symptoms and physical exam: fever, nasal crusting, hearing loss (especially in AAV), uveitis (hypocomplementemic urticarial vasculitis syndrome), arthritis (connective tissue diseases), myalgia with or without consequent severe limitation in movements (connective tissue diseases, AAV, PAN). Symptoms of connective tissue diseases should be noted, such as dry eyes or mouth, photosensitivity and/or facial cutaneous eruption, oral lesions or genital lesions (aphthous stomatitis, ulcers), or muscle weakness. Past medical history, including hospitalizations, underlying diseases/disorders (including febrile diseases), pre-immunization signs and symptoms including identification of indicators for, or the absence of, a history of allergy to vaccines, vaccine components or any medications; food allergy; allergic rhinitis; atopic eczema; asthma, autoimmune diseases.10 | |
| Laboratorial tests and other exams: marked elevation of erythrocyte sedimentation rate and c-reactive protein, while muscle enzymes were generally normal (PAN). Electromyography and/or MRI in suspect of myositis. Muscle biopsies, mostly revealing a histological pattern compatible with PAN, presenting with necrotizing arteritis, leukocytoclastic angiitis, fibrinoid necrosis of small and medium arteries and leukocyte infiltration of the vessel walls or perivasculitis in systemic PAN. Complement serum protein consumption is suggestive of systemic vasculitis as connective autoimmune tissue diseases (SLE, rheumatoid arthritis, Sjögren syndrome), cryoglobulinemic vasculitis, malignancy-associated vasculitis and septic vasculitis. | ||
| Etiologic associated factors | Drugs, non-prescribed medicines and immunizations | Several medications, as penicillin and derivates, sulfonamides, quinolones, hydantoins, insulin, tamoxifen, oral contraceptives, phenothiazines, allopurinol, thiazides, retinoids, radiocontrast agents and anti-influenza vaccine. Chemical substances (e.g., insecticides and petroleum products) and foods (e.g., milk proteins and gluten) can also cause a vasculitis. Many illicit drugs can cause vasculitis, with the best evidence being that for the sympathomimetic drugs. Vitamins and nutritional supplements have also been reported to cause vasculitis. Drug-Induced Vasculitis (DIV) is a diagnosis of exclusion, as there are no laboratory studies diagnostic for DIV. Although eosinophilia is common in systemic DIV (79%), it is present in only 22% of DIV limited to the skin.13 Drug-induced AAV may be associated with: propylthiouracil, hydralazine, minocycline, anti-TNF alfa agents, acitretin, allopurinol, methimazole, and other. |
| Infections and parasitic infestations | Infections can cause lesions which both mimic vasculitis as well as those which represent either a septic or true reactive vasculitis. Infection can present in many ways, including septic vasculitis, upper respiratory tract flares of GPA, PAN, cryoglobulinemic vasculitis, urticarial vasculitis, IgA-vasculitis, and cutaneous small-vessel vasculitis (CSVV, a SOV).13 Common agents include bacteria, viruses, parasites, and fungi. Special mention should be given to the known association of hepatitis B virus with PAN and hepatitis C virus with mixed cryoglobulinemia.13 Other complementary exams should include an investigation for asymptomatic or oligosymptomatic infections and/or parasite infestations as anti-streptolysin O, tuberculosis (Mantoux test and/or QuantiFERON-TB Gold In-Tube assay), Parvovirus B19 (serology IgM and IgG), schistosomiasis (eggs in stool exam), toxocariasis (serology IgG, peripheral eosinophilia), strongyloidiasis (larvae in stool exam and serology) and syphilis (serology and/or immunohistochemistry in cutaneous biopsy). Others agents are reported in case reports or case series as, (i) Bacterial agents: Streptococcus (Streptococcus viridans; group A, C, and G streptococci), enterococci, Staphylococcus aureus, Mycobacterium leprae (Lucio's phenomenon in leprosy and vasculitis in erythema nodosum), Salmonella species, Borrelia burgdorferi, Pseudomonas aeruginosa, Legionella pneumophila; (ii) Parasites: Schistosoma haematobium, Onchocerca volvulus, Cysticercus, Loa loa; (iii) Viruses: HIV infection, herpes simplex 1 and 2, Cytomegalovirus, Epstein–Barr, Varicella-Zoster, Rubella, adenovirus, echovirus, coxsackievirus, parainfluenza, hepatitis A virus infections, and (iv) Fungi: Aspergillus spp., Fusarium spp., Coccidioides immitis.14 | |
| Inflammatory systemic conditions (autoimmune diseases) | SLE, Sjögren's syndrome, rheumatoid arthritis, scleroderma, dermatomyositis, complement deficiency, alpha1 antitrypsin deficiency, inflammatory bowel disease, chronic active hepatitis, intestinal bypass surgery, primary biliary cirrhosis, relapsing polychondritis, and other. | |
| Neoplasia and paraneoplastic syndrome | Hematologic disorders constitute the most common group of malignancies associated with cutaneous vasculitis (lymphoma Hodgkin and non-Hodgkin, acute myelogenous leukemia, chronic lymphoid or lymphocytic leukemia, lymphoblastic leukemia, immunoblastic sarcoma, megakaryocytic leukemia malignant B-cell lymphoma, T-cell lymphoma, myelodysplastic syndrome, myelofibrosis) and less commonly the vasculitis related to solid organ tumors (colon carcinoma, renal carcinoma, colon adenocarcinoma, prostate carcinoma, lung carcinoma, lung squamous carcinoma, gastric tubular carcinoma, gastric adenocarcinoma, pancreatic carcinoma, breast carcinoma, ovarian cancer, pharyngeal carcinoma, vocal cord carcinoma, pelvic carcinoma, hepatocarcinoma, urinary bladder carcinoma).15 Cutaneous paraneoplastic vasculitis is an entity not uncommonly encountered by clinicians. In these cases, the presence of cytopenia and immature cells may be red flags for the diagnosis of cancer. In patients with paraneoplastic cutaneous vasculitis, the prognosis depends on the underlying neoplasia.15 | |
| Idiopathic | Despite the efforts of the clinicians on work-up for an etiology to cutaneous or systemic vasculitis, most of patients remain as undetermined etiology. Some authors refer an idiopathic origin for vasculitis in 45–50%16 to 6917–79%18 of the patients suffering of CSVV. In both CSVV and IgA vasculitis, the most common etiologic association is idiopathic. |
AAV, ANCA-Positive Vasculitis; CNS, Central Nervous System; CSVV, Cutaneous Small-Vessel Vasculitis; CT, Computed Tomography; DIV, Drug-Induced Vasculitis; EGPA, Eosinophilic Granulomatosis with Polyangiitis; GPA, Granulomatosis with Polyangiitis; MPA, Microscopic Polyangiitis; MRI, Magnetic Resonance Image; PAN, Systemic Polyarteritis Nodosa; PET, Positron Emission Tomography; PNS, Peripheral Nervous System; SLE, Systemic Lupus Erythematous; SOV, Single Organ Vasculitis of the skin.
Some clues may help to recognize both the size of the vessel involved and the suspect diagnosis. Arterial hypertension may suggest medium-vessel vasculitis of the renal vasculature, as seen in systemic PAN. Cutaneous lesions as palpable purpura (the most frequent type of lesion in CSVV), nonpalpable purpura, pinpoint papules, vesicle, pustules, splinter hemorrhages, and urticaria are often suggestive of small vessel vasculitis. Vesicles and bullous lesions often arise on purpuric macules. A subsequent hemorrhagic pattern is observed, and these lesions may produce ulcerations in the skin. Nonpalpable purpura on the lower extremities can be found in patients with Sjogren syndrome and other conditions that include hypergammaglobulinemic purpura, cryoglobulinemic vasculitis and Waldenström's hypergammaglobulinemic purpura. Urticarial lesions can occur in Systemic Lupus Erythematosus (SLE), Sjogren syndrome, hypocomplementemic urticarial vasculitis syndrome, and EGPA (Fig. 2).13
 Purpura, petechiae, vesicles and hemorrhagic blisters in the lower limbs in a patient with cryoglobulinemic vasculitis, (b) Urticated sometimes confluent lesions and purpura in the lower limbs very suggestive of urticaria vasculitis.")
Different lesions usually seen in patients with small vessel vasculitis. (a) Purpura, petechiae, vesicles and hemorrhagic blisters in the lower limbs in a patient with cryoglobulinemic vasculitis, (b) Urticated sometimes confluent lesions and purpura in the lower limbs very suggestive of urticaria vasculitis.
In medium-sized vessels vasculitides, patients often present with subcutaneous nodules, livedo, large or deep ulcers, papulonecrotic lesions, and digital infarcts as indicative involvement of occlusive or semi-occlusive of medium vessels vasculature (vascular plexus of dermo-hypodermal junction or exclusively of the subcutaneous tissue). Subcutaneous nodules surrounded by a livedoid pattern are more characteristic of Polyarteritis Nodosa (PAN), cutaneous arteritis, sarcoidosis vasculitis, or SLE, rather than Antineutrophil Cytoplasmic Antibodies (ANCA)-Associated Vasculitides (AAV) (Figs. 3 and 4).13–19
 Subcutaneous nodules and ulcers in a patients with cutaneous arteritis, (b) Extensive ulcers with areas of necrosis and residual atrophic scars in the lower limbs of a patients with microscopic polyangiitis.")
Distinct lesions normally found on the skin of patients with medium vessel vasculitis: (a) Subcutaneous nodules and ulcers in a patients with cutaneous arteritis, (b) Extensive ulcers with areas of necrosis and residual atrophic scars in the lower limbs of a patients with microscopic polyangiitis.
 Livedo racemosa in the lower limbs, including the dorsum of the feet in a patient with cutaneous arteritis, (b) Digital necrosis in a patients with ANCA-associated vasculitis - granulomatosis with polyangiitis.")
Distinct lesions normally found on the skin of patients with medium vessel vasculitis: (a) Livedo racemosa in the lower limbs, including the dorsum of the feet in a patient with cutaneous arteritis, (b) Digital necrosis in a patients with ANCA-associated vasculitis - granulomatosis with polyangiitis.
Cutaneous lesions of Winkelmann granulomas are characterized by the occurrence of tender, erythematous to violaceous papulonodules, usually symmetrically distributed on the elbows or distal upper extremities and fingers. Rarely, subcutaneous indurated cord-like bands (‘rope sign’) have been observed. Histologically, this disease is a variant of CLA with prominent influx of neutrophils with palisading macrophages surrounding a central necrosis composed of basophilic fibrin, degenerative collagen and, of course, neutrophils and neutrophilic debris. However often a true vasculitis is not observed in histopathology sections. This skin reaction pattern was originally described in EGPA disease but subsequently observed in a variety of infectious diseases such as subacute bacterial endocarditis, hepatitis, lymphoproliferative and autoimmune disorders such as GPA, rheumatoid arthritis, inflammatory bowel disease, Takayasu's arteritis and (flaring) SLE.20
Digital infarcts are commonly seen in vasculitis associated with rheumatoid arthritis but are also seen in PAN and AAV (Fig. 5a).21 Retiform purpura in plaques (Fig. 5b) is an uncommon finding of palpable purpura in a livedoid, reticulate or arciform pattern. This arrange is related to the physiological vascular anatomy and results from ischemia-related hemorrhage around a dermal/subcutaneous vessel prior to complete occlusion. Although retiform purpura may occur in a variety of clinical settings, retiform purpura in plaques indicates vascular inflammation and more distinct pathophysiology, as well non-inflammatory conditions, without a true vasculitis, didactically grouped as cutaneous pseudovasculitis (antiphospholipid syndrome, calciphylaxis, warfarin-induced cutaneous necrosis, heparin-induced skin necrosis, cryoglobulinemia, cholesterol or crystal emboli, and oxalosis).21
-induced antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis: (a) Palpable purpura in a livedoid, reticulate pattern with a necrotic center located in the tighs. The recognition of a retiform purpura pattern indicates that a medium vessel component of vasculitis exists, (b) Large ulcerations with irregular borders, granulating wound bed, areas of necrosis and eschar, and serous discharge.")
Retiform purpura in a patient with Propylthiouracil (PTU)-induced antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis: (a) Palpable purpura in a livedoid, reticulate pattern with a necrotic center located in the tighs. The recognition of a retiform purpura pattern indicates that a medium vessel component of vasculitis exists, (b) Large ulcerations with irregular borders, granulating wound bed, areas of necrosis and eschar, and serous discharge.
A general panel of complementary exams suggested by our study group includes several tests, previously cited in table 3 and other, especially for those patients without an obvious cause of vasculitis: complete blood cell count with differential, blood urea nitrogen/creatinine, liver function panel, urinalysis, stool guaiac, serum protein electrophoresis with immunofixation to search for evidence of a paraproteinemia, antistreptolysin-O, HBV/HCV/HIV serologies, cryoglobulins, complement levels (CH50, C3, C4), antinuclear antibody, ANCAs and Rheumatoid Factor (RF).9,13 If the patient is febrile and has a heart murmur, then blood cultures and echocardiography should be performed. For cases with a medium-sized vessel involvement or signs and symptoms of connective tissue disease, an Antinuclear antibody (ANA) should be obtained.13
C-reactive protein levels usually reflect AAV activity. In both AAV and PAN anti-streptolysin O titers should be evaluated because of the common role of streptococci in these settings. Chest radiology studies, gastrointestinal image exams, gynecological examination for females and urological evaluation form male patients are an adequate screening for malignancy in the absence of any other specific signs or symptoms. High fever, weight loss, bicytopenia or severe anemia, Raynaud's phenomenon (in the absence of connective tissue disease), or cryoglobulins should prompt a more thorough search for malignancy.13
Since only a few vasculitides syndromes have pathognomonic clinical, radiographic and or laboratory findings confident and accurate diagnosis requires a representative skin biopsy to histological confirmation. On the other hand, a biopsy diagnosis of vasculitis cannot stand alone, as it must be correlated with clinical history, physical and laboratory findings and/or angiographic features. For example, a diagnosis of SOCV requires that no evidence of systemic manifestations is found.10 If systemic vasculitis is suspected, imaging studies can provide a useful means to determine disease extent and activity, and serology, such as C-reactive protein, erythrocyte sedimentation rate and ANCA levels, and type, can be used to monitor disease activity and predict mortality risk, respectively.
Hence, the classification of cutaneous vasculitis into specific syndromes is better approached morphologically by determining vessel size and main inflammatory response. These histological patterns (Table 2) combined with Direct Immunofluorescent (DIF) examination, ANCA status and findings from work-up for systemic disease, allow for specific diagnosis, and ultimately, a more effective therapy. A neutrophil-predominant small vessel vasculitis primarily affecting upper dermal blood vessels and showing papillary IgA deposits is diagnostic of IgA-vasculitis, whereas a neutrophil-predominant small vessel vasculitis affecting dermal and subcutaneous blood vessels with predominance of IgM vascular deposits would implicate Cryoglobulinemic Vasculitis (CV) or Rheumatoid Vasculitis (RV).22
Clinicians more trained in recognizing elementary cutaneous lesions must be able to perform a representative biopsy of the necessary skin layers. In general, a deep and large punch biopsy (5–6mm in diameter) or an excision biopsy is the preferred mean to sample vessels of all sizes.22 Some key points to a proper cutaneous biopsy are listed below:
- a)
A sample biopsy extending to subcutis taken from the most tender, reddish or purpuric, lesioned skin is the key to obtaining a significant diagnostic result;
- b)
The optimal time for skin biopsy is <48h after the appearance of a vasculitis lesion. If the biopsy is poorly timed, the pathological features of vasculitis may be absent, a fact that must be considered when interpreting a negative biopsy from a patient whose clinical findings suggest vasculitis;
- c)
Purpuric lesions biopsied in the first 24h display fibrin deposits within the vessel wall accompanied by neutrophilic infiltration of the wall and surrounding hemorrhage and nuclear debris (leukocytoclasia). After 24h, neutrophils begin to be replaced by lymphocytes and macrophages. Thus, biopsy of lesions >48h old, regardless of the underlying form of vasculitis, may show lymphocyte-rich infiltrates, troubling the clinical syndromic vasculitides diagnosis;
- d)
Biopsy specimens should be obtained from non-ulcerated skin lesions due to the frequent finding of incidental vasculitis underlying an ulcer bed. If only superficial ulcers are present, biopsy of the edge is acceptable. In the case of deep, ‘punched out’ ulcers, biopsy of the subcutis including the central ulcerated area increases diagnostic yield and recognition of an arterial vasculitis, such as systemic PAN or cutaneous arteritis;
- e)
In case of livedo racemosa, a deep biopsy specimen should be taken from the center of the circular livedo segment because it is where is located the stenosed vessel responsible for the cyanotic periphery. In this setting, level sections are often required to find the focus of vasculitis, which is typically focal and segmental.
Mimouni et at.23 reviewed 29 consecutive patients presenting white atrophy, 6 of whom had underlying medium-sized vasculitis consistent with PAN. Three of them had been previously diagnosed as having livedoid vasculopathy on superficial biopsies. So, particularly in the setting of white atrophy repeated and deep biopsies are often necessary to reveal the accurate underlying pathology of necrotizing medium-sized vasculitis in the reticular dermis and the subcutis. Meanwhile, Wohlrab et al.24 evaluated the sensitivity of skin biopsies in Sneddon's syndrome. The authors collected 5 deep-punch biopsies (4mm at least) from different areas where livedo was detected (3 from white and 2 from red areas) in 15 patients. The method had a sensitivity of 27% with one biopsy, 53% with two biopsies and 80% with three biopsies taken from white areas in all cases.
DIF studies should be performed whenever it is possible. For example, the diagnosis of IgA-vasculitis can only be made when IgA vascular deposits are found.22 In cases of immune complex-mediated vasculitis, 100% of skin samples will reveal immunoglobulins within the first 48h, as 70% will do it at 48–72h. After this interval, despite complement deposits may still be detected in more than 50% of vasculitis skin lesions, immunoglobulins will not.22
Our group performed a retrospective study on the DIF results in a tertiary center in Brazil from January 2007 to December 2014. We evaluated 235 cutaneous samples in 282 patients with CLA. Age ranged from 5 to 87 years-old, with a median age of 45 years. 191/282 (67.73%) of the patients were female. DIF analysis showed positivity in 70.21% of the samples, and C3 was the most frequent immunoreactant. IgA deposition at the blood vessel wall was related to age and absence of autoimmune/inflammatory diseases, while Immunoglobulin M (IgM) deposition was related to female gender, autoimmune/inflammatory disorders, C3 and C4 consumption, and antinuclear antibody or anti-SSA/anti-SSB positivity. Immunoglobulin G (IgG) deposition at the blood vessel wall was associated with age and positive ANCA. Finally, C3 deposition at the blood vessel wall, the most frequent finding of this study, was associated with hematuria and renal involvement. Systemic involvement was present in 12.5% of patients with CLA.17
In summary, in cases of cutaneous vasculitides it is essential to select the proper site of cutaneous biopsy. The histopathological features of CLA may include neutrophils invading the vessel wall, fibrinoid necrosis or fibrin within the vessel wall, nuclear dust (leukocytoclasia), extravasated erythrocytes, perivascular inflammatory cell infiltration, mainly neutrophils, and endothelial swelling. Biopsies for DIF should be taken in the first 8–24h of appearance of the lesion. Otherwise, the inflammation destroys the immune complexes and they become falsely negative. Detection of immune complex deposition may have diagnostic and prognostic significance. Besides immunoreactants in the vessel wall, DIF can reveal deposition of IgG and C3 at the dermo-epidermal junction, pointing out a positive lupus band test, or may be related to hypocomplementemic urticarial vasculitis with an underlying LES.22
Epidemiology data on vasculitisThe annual incidence of CLA is estimated in 45/million in the United States (population-based in Olmsted County, Minnesota – cutaneous biopsy-proven cases).18 It affects both genders equally and patients of all ages.9 In children, by contrast, IgA-vasculitis is much more common than non-IgA small vessel vasculitis of the skin.9 The presence of an underlying systemic vasculitis, connective tissue disease, or malignancy is much more common in adults than in children.9,18
In a review on systemic vasculitides, Elefante et al.25 presented some data about the epidemiology of AAV: (i) ANCA specificity was associated with different genetic background, clinical features, treatment response, and prognosis in terms of relapse rate and survival rate; (ii) distribution of ANCA specificity differs significantly between the ethnic groups; (iii) MPO-ANCA resulted significantly more frequent in Asian populations [Japanese OR=59.2 (95% CI 8.0–440.7), p<0.001, Chinese OR=6.8 (95% CI 2.6–17.8), p<0.001], Caucasian American [OR=2.6 (95% CI 1.7–4.0), p<0.001] and Middle Eastern/Turkish [OR=2.3 (95% CI 1.3–4.2), p<0.005], when compared to the Northern Europeans, who presented more PR3-ANCA specificity; (iv) ANCA negativity was also significantly more frequent in Caucasian Americans than Northern Europeans [OR=2.0 (95% CI 1.3–3.2), p=0.002], and (v) there are differences in frequency of organ involvement between the ethnic groups, when compared to Northern Europeans, the Asian population presented significantly less ocular and ear, nose, and throat involvement while renal involvement was significantly less frequent in Caucasian Americans and significantly more frequent in Middle Eastern/Turkish, however interestingly, some differences were not completely determined by the differences in ANCA pattern.
PathogenesisWe can simplify the mechanisms involved in the pathogenesis of vasculitides into two pathways: (i) immune-complex associated vasculitides and (ii) AAV. However, several factors are involved as: genetic basis, innate and acquired immunity, pathogens, immune tolerance disruption, and “autoantigens”.
The causes of CSVV include infections (20%), inflammatory conditions (15–20%), drug reactions (10–15%), and malignancies (5%).26 Disease-inducing or promoting factors are not known for more than half of the cases, and so they are currently classified as idiopathic.10 Although non-immunologic factors such as direct infection of endothelial cells can cause vasculitis, most lesions are mediated by immunopathogenic mechanisms. These mechanisms can be classified into Gell and Coombs’ four types of hypersensitivity reactions. The majority of cutaneous lesions are likely due to type III hypersensitivity reactions. Immune complexes deposition in postcapillary venules activates complement, which, in turn, induces mast cell degranulation and neutrophil chemotaxis.10
Virtually all drugs have been considered as potential precipitating agents for vasculitis and CSVV is by far the most common form of drug-associated vasculitis. According to the study of Ortiz-Sanjuán et al.27 the most frequently associated were antibiotics, mainly B-lactams and quinolones. Other agents involved were nonsteroidal anti-inflammatory drugs, paracetamol, allopurinol and anticonvulsants. In this series of 239 cases, patients with vasculitis due to major infections, such as endocarditis or pneumonia, were excluded.
RV is probably the most widely recognized form of secondary vasculitis and is typically seen in long standing, erosive, seropositive rheumatoid arthritis. Deposition of immune complexes and subsequent activation of the complement cascade is believed to play a key role in the pathogenesis of RV. An increased expression of inflammatory cytokines such as TNF-α, IL-1 and IL-6 is noted, although their exact role in the pathophysiology of RV remains unclear. Meanwhile, cutaneous vasculitis may be seen in approximately 19–28% of patients with SLE.28 Small vessels especially post capillary venules are involved in most cases (80–90%).28 Presence of anti-endothelial antibody is detected in up to 80% of these patients and might contribute by activation of endothelial cells, direct cytotoxic effect due to complement-dependent cytotoxicity or indirect cytotoxic effect secondary to antibody-dependent cytotoxicity.28
Genetic susceptibility for vasculitidesGenome-wide association studies have revealed a pivotal role of the Human Leukocyte Antigen (HLA) region in the genetic susceptibility to vasculitides. However, each form has distinct HLA association markers that define them, most likely due to disease-specific differences in antigenic drivers. Despite the considerable advance in the identification of consistent genetic risk factors during the last 10 years, the number of identified risk loci for most types of vasculitides remains significantly lower than other immune-mediated diseases.29
There is a strong association with HLA class II alleles with IgA-vasculitis pathogenesis, specifically HLA-DRB1 in Europeans, mainly due to HLA-DR1*0103.43, suggesting that it may be related to other class II vasculitides such as giant cell arteritis or AAV. It has also been proposed that, although less strong, there is a potential effect of HLA class I in the pathogenesis of IgA vasculitis. When it comes to AAV, the genetic distinctions within this group may be related to the antigenic specificity of ANCA rather than to the clinical syndrome. Different AAV subtypes are underpinned by distinct risk factors, with GPA being associated with HLA-DP, SERPINA1, PRTN3 and semaphorin 6A, whereas MPA is mainly associated with HLA-DQ polymorphisms.29–31
Takayasu arteritis is predominantly related to HLA class I alleles, in particular to HLA-B52, as it is Behçet's Disease (BD).30 The strongest single-risk factor for BD was confirmed to be HLA-B51, but other HLA-B alleles with lower independent effects were also observed, for example, HLA-B15 and HLA-B27.29 Outside the major histocompatibility complex region, susceptibility to BD has been associated to interleukin 10 and the region of interleukin 23 receptor/interleukin 12 receptor β2.29
The association of HLA-DR alleles with PAN has been reported, but with variable results according with the studied population. Unfortunately, there is little information on genetic susceptibility to hypersensitivity vasculitis (CSVV or SCOV). Genetic studies on this vasculitis are scarce and no association of this condition with the HLA region was confirmed, suggesting a heterogeneous etiology in the pathogenesis of this vasculitis that often is restricted and limited to skin.30
Infections as a trigger of vasculitidesVarious immune mechanisms are involved in infection-associated vasculitides pathogenesis. Most of them are caused by direct invasion and proliferation of pathogens in the vascular walls causing inflammation. On the other hand, they can sometimes be attributed to indirect immunological effects such as type II, III, or IV hypersensitivity reactions triggered by the infection. Structures such as fimbriae, besides MSCRAMMs (microbial surface components that recognize adhesive matrix molecules) and others cell-wall-anchored proteins are utilized by bacteria to make their way into vulnerable cells. It has been demonstrated that viruses have analogous membrane-adhesion molecules. Nonetheless, whether their density of expression has a role in determining the frequency with which distinct organs are affected is a knowledge to be seek in scientific vasculitides-research related.2,32
Vasculitis has been reported in association with numerous microorganisms, ranging from viruses to fungi. In specific situations, the relationship between etiology and angiocentric inflammation and destruction is clear, as it is in aortitis caused by Mycobacterium tuberculosis or by Treponema pallidum, for which there is a predilection for the ascending aorta. However, other infections, such as HIV, are associated with a wide variety of vasculitic phenotypes, affecting small, medium or large vessels. In these cases, necrotizing, non-necrotizing, giant cell and eosinophilic arteritis have been observed at histology. Direct vessel wall invasion by HIV itself or by opportunistic organisms, and immune complexes containing HIV antigens and antibodies are some of the mechanisms of injure proposed.33
In fact, several infectious diseases are implicated in the production of immune-complex-mediated forms of vasculitis.2 IgA vasculitis has been reported to occur in association with various bacteria, including Mycoplasma pneumoniae and Clostridium difficile. As Ureaplasma urealyticum, Aerococcus viridians, Burkholderia cepacia and Listeria monocytogenes have been incriminated in CLA.33 Although the incidence of PAN has significantly decreased in the prevailing scenario of the vaccination programs and blood transfusion screening, more than 30% of the cases are secondary to HBV infection. Most evidence supports a type III hypersensitivity injury whereby vascular damage is produced by immune complexes with viral antigens. There have been anecdotal reports of chronic HBV infection associated with other forms of vasculitides such as GPA, even though it still remains unclear whether these reports are causal or coincidental.32
The pathogenesis of HCV-associated vasculitis seems to involve a direct interaction between the virus and lymphocytes leading to polyclonal activation and proliferation of B cells producing IgM with RF activity.33 IgM RF is capable of activating the complement system through the binding of the globular domain of the C1qprotein. C1q receptors (gC1q-R) are widely expressed on the surface of blood cells and endothelial cells, and they link to large immunocomplexes containing HCV core protein, facilitating subsequent vascular inflammation. The immunocomplexes with cryoprecipitate containing viral antigens, IgM RF bound to polyclonal anti-HCV IgG, and complement are localized to small vessels of internal organs, although preferentially to the colder extremities.32
HCV-associated CV is a systemic small-to-medium vessel vasculitis due to vascular deposition of cold-precipitable serum proteins, called cryoglobulins. Type I cryoglobulins are monoclonal immunoglobulins, type II cryoglobulins consist of monoclonal immunoglobulins with a RF activity, associated with polyclonal IgG, whereas type III cryoglobulins comprise polyclonal IgG and IgM with RF activity. Elefante et al.25 summarized the clinical, laboratorial and predisposing factors influencing the outcome in patients with CV unrelated to HCV: (i) essential (no identifiable underlying disease) CV was the largest group (39.4%) and the first associated condition (21.1%) was primary Sjogren's syndrome (pSS); (ii) overt purpura was present in 78% of patients of pSS group, 64% of whom had type II cryoglobulins, and in patients with pSS the presence of cryoglobulins was associated with highest systemic activity, suggesting that all patients with pSS should be tested for serum cryoglobulins at least at the time of their diagnosis; (iii) SLE-related CV was present in 10.9% of cases as well as other immune conditions, HBsAg positivity in 8.6%, lymphoproliferative disease in 6.8% and some solid tumors in 2.3%; (iv) type II cryoglobulins were present in 54.9% and were independently associated with purpura and fatigue, and (v) older age, male gender, type II cryoglobulins and HBsAg were independently associated with great mortality in patients with HCV-unrelated CV.
PAN is divided into 2 subtypes: systemic and cutaneous. It predominantly presents between 40 and 60 years of age. Besides HBV infection, systemic PAN has been linked to other infectious agents including HCV, HIV, cytomegalovirus, parvovirus B19, and HTLV. The cutaneous PAN has been attributed to streptococcal infections in the pediatric population.26
ANCA-associated vasculitidesThe ANCAs are a group of autoantibodies, mainly of the IgG type, developed and released from B cells against antigens in the cytoplasm of neutrophil granulocytes. One type of ANCA is associated with diffuse staining of the cytoplasm and is known as cytoplasmic ANCA (c-ANCA), whilst the other is related with staining around the nucleus and is known as perinuclear ANCA (p-ANCA). The major antigen targeted by c-ANCA is PR3 and that targeted by p-ANCA is MPO. Both are expressed on the cell surface of neutrophils activated by pro-inflammatory cytokines such as IL-1β and TNF generated during infections. ANCAs link to these antigens. Meanwhile, the Crystallizable Fragment (Fc) portion of these ANCAs binds to Fcγ receptors on neutrophils, inducing their excessive activation. This fact leads to abnormal cytokine production, release of Reactive Oxygen Species (ROS) and lytic enzymes, and eventually Neutrophil Extracellular Traps (NET) formation.34,35
AAV constitutes a group of rare diseases characterized by necrotizing inflammation of small to medium-sized blood vessels and the presence of ANCA. They are represented by GPA, MPA with its renal limited form pauci-immune necrotizing crescentic glomerulonephritis and EGPA. Results from studies have demonstrated that ANCA, detected by the immunofluorescence technique (c-ANCA or p-ANCA), are a sensitive marker for the so-called AAV, with sensitivity ranging from 80% to more than 90%. Unfortunately, immunofluorescence has a low specificity (80% or less), which is mainly caused by positive p-ANCA in disease controls, such as in inflammatory bowel diseases. P-ANCA, in disease controls, can be also caused by the presence of anti-nuclear antibodies. Antigen specificity (PR3 or MPO) does not effectively differentiate among the AAV, however c-ANCA/PR3-ANCA are mainly found in GPA, while p-ANCA/MPO-ANCA are more prevalent in MPA, pauci-immune necrotizing crescentic glomerulonephritis and CSS. ANCA are detected in 70%–90% of active, generalized GPA, but only in about 40%–50% of the loco-regional forms.36 Beyond a diagnostic serological marker, data support a pathogenic role for ANCA, which promote activation of primed neutrophils and monocytes, and their adhesion to the endothelium, leading to subsequent tissue damage.19
The proteins of complement system participate in the pathogenesis of several small-medium vessels vasculitis including AAV, CV, IgA vasculitis and urticarial vasculitis. PR3- and MPO-ANCA-activated neutrophils prompt C5a release. Subsequent interaction of C5a (a protein derivate from alternative complement pathway activation) with the C5a receptor 1 (C5aR1) may represent a proinflammatory amplification loop in AAV.30 Due this complement activation, elevated serum and plasma concentrations of C5a and C3a have been observed in active AAV, especially MPO-ANCA positive vasculitis.31
Besides microbial components such as peptides or lipopolysaccharides and complement, certain drugs mainly propylthiouracil (PTU), hydralazine and levamisole-adulterated cocaine stimulate ANCA autoantigen expression on the membrane surface of neutrophils. Although these immune cells are the paramount player in the pathogenesis of AAV, since they are both effector cells responsible for endothelial damage, and targets of autoimmunity, it has been demonstrated that some others including CD4 T cells and monocytes are also involved.35
Neutrophil extracellular traps (NETs) and vasculitisNeutrophils are able to generate NETs, which are found in a variety of conditions, such as infection, malignancy, atherosclerosis, and as previously mentioned autoimmune diseases, such as AAV. Although several stimuli are responsible for neutrophil activation and NETs formation, a little is known on the exact pathway leading to induction of NET formation.37 The strongest evidence that NETs actually serve as a source of autoantigens driving autoantibody production in vasculitis comes from studies on drug-induced vasculitis. MPO-ANCA positivity is relatively common in patients treated with propylthiouracil, and some of these patients develop a vasculitis-like syndrome.38 MPO and PR3 have been demonstrated within NETs isolated from patients with AAV, and ANCA can further induce NET formation in these patients. ANCA itself also can induce autophagy of neutrophils, which promotes NETosis, a unique form of programmed cell death.38
NETs have been shown to be present not only in skin lesions and thrombi from AAV patients, but also in the circulation. NETs containing pro-inflammatory proteins are thought to contribute to vasculitis by direct damage to endothelial cells, activation of the complement system (especially alternative complement pathway), generating p/c-ANCA, and thrombosis formation.37 However, Wang et al.39 demonstrated that circulating levels of NETs cannot be used as a biomarker to assess disease activity in AAV patients.
ConclusionA great number of significant contributions have been made on the classification, pathogenesis, clinical sub-setting of cutaneous and systemic vasculitides in this paper. The chief reason for the ongoing research is to strive a personalized medicine based on endotypes rather than phenotypes. The capacity of recognizing integumentary lesions of different vasculitides, allied to other systemic symptoms and signs, an adequate biopsy and proper complementary laboratory and image exams can contribute to the correct diagnosis of this challenging group of diseases.
Financial supportThis study was carried out with support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Código de Financiamento 001.
Authors’ contributionsThâmara Cristiane Alves Batista Morita: Elaboration and writing of the manuscript; intellectual participation in the propaedeutic and/or therapeutic conduct of the studied cases; critical review of the manuscript.
Gabriela Franco S. Trés: Approval of the final version of the manuscript; elaboration and writing of the manuscript; intellectual participation in the propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; critical review of the manuscript.
Roberta Fachini Jardim Criado: Approval of the final version of the manuscript; conception and planning of the study; elaboration and writing of the manuscript; effective participation in research orientation; critical review of the literature; critical review of the manuscript.
Mirian Nacagami Sotto: Critical review of the manuscript.
Paulo Ricardo Criado: Approval of the final version of the manuscript; conception and planning of the study; elaboration and writing of the manuscript; obtaining, analysis, and interpretation of the data; effective participation in research orientation; intellectual participation in the propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; critical review of the manuscript.
Conflicts of interestNone declared.
How to cite this: Morita TCAB, Trés GFS, Criado RFJ, Sotto MN, Criado PR. Update on vasculitis: an overview and dermatological clues for clinical and histopathological diagnosis – Part I. An Bras Dermatol. 2020;95:352–68.
Study conducted at the Department of Dermatology, Faculdade de Medicina, Universidade de São Paulo and Department of Dermatology, Faculdade de Medicina do ABC, Santo André, SP, Brazil.
