










Úlceras isquêmicas por comprometimento da microcirculação dos membros inferiores são causas de úlceras dolorosas que oferecem desafio para o diagnóstico correto e tratamento. Vasculopatia livedoide, calcifilaxia e úlcera hipertensiva de Martorell fazem parte desse grupo e apresentam algumas semelhanças entre si em virtude do comprometimento oclusivo microvascular. São frequentemente diagnosticadas erroneamente como úlceras inflamatórias, do tipo pioderma gangrenoso, e vasculites. Nesta revisão são discutidos fisiopatogenia, fatores de risco, aspectos clínicos, diagnósticos diferenciais, histopatologia e apresentada uma atualização terapêutica da vasculopatia livedoide, calcifilaxia e úlcera de Martorell. Embora sejam causas menos frequentes de úlceras crônicas, o diagnóstico correto é fundamental para diminuir a chance de terapêuticas equivocadas que podem impactar na morbimortalidade relacionadas a essas condições.
As úlceras crônicas de membros inferiores são condições comuns entre adultos, com inúmeras causas, e muitas vezes representam desafio diagnóstico e terapêutico. As principais causas das úlceras crônicas de membros inferiores são doenças venosas, arteriais e neuropáticas.1 As úlceras isquêmicas, como as de origem arterial, desenvolvem‐se em decorrência de suprimento sanguíneo inadequado por comprometimento da macrocirculação arterial. Sua causa mais comum é a doença aterosclerótica.2 Entretanto, neste artigo, abordaremos as outras causas de úlceras isquêmicas dos membros inferiores, relacionadas ao comprometimento oclusivo da microcirculação; são elas: vasculopatia livedoide, calcifilaxia e úlcera isquêmica hipertensiva. Embora sejam causas menos comuns de úlceras crônicas de membros inferiores, elas devem ser reconhecidas clinicamente e fazer parte dos diagnósticos diferenciais, pois geralmente são confundidas com outras causas, o que ocasiona atraso no diagnóstico e no tratamento.3
Vasculopatia livedoideDefiniçãoA vasculopatia livedoide (VL) é doença ulcerativa crônica com curso clínico marcado por períodos de remissões e exacerbações. A VL acomete a região distal das pernas, os tornozelos e o dorso dos pés (fig. 1).4 O quadro é intensamente doloroso e provoca significante redução da qualidade de vida.5 A VL é doença trombótica distinta da vasculite, pois não é caracterizada primariamente por danos na parede dos vasos ou infiltrado inflamatório. Alguns autores não a consideram entidade única, mas a manifestação cutânea de diversas doenças que cursam com hipercoagulabilidade, trombose dos vasos da derme e redução da fibrinólise, como doenças autoimunes, síndromes neoplásicas, anomalias congênitas ou adquiridas do sistema fibrinolítico, dentre outras.6,7
 Lesões ativas caracterizadas por pequenas úlceras com crostas e borda eritemato‐purpúricas localizadas na face medial das pernas e pés. Nota‐se também livedo racemoso no dorso dos pés. (B) Lesões em remissão na face lateral da perna e pé com cicatrizes acastanhadas e estelares branco‐marfínicas caracterizando a atrofia branca de Millian.")
Vasculopatia livedoide. (A) Lesões ativas caracterizadas por pequenas úlceras com crostas e borda eritemato‐purpúricas localizadas na face medial das pernas e pés. Nota‐se também livedo racemoso no dorso dos pés. (B) Lesões em remissão na face lateral da perna e pé com cicatrizes acastanhadas e estelares branco‐marfínicas caracterizando a atrofia branca de Millian.
A VL é considerada doença rara com incidência estimada em 1:100.000 por ano. Predomina nas mulheres, em razão de 3:1. As lesões podem se agravar nos meses mais quentes do ano.7 A faixa etária mais acometida é dos 15 aos 50 anos, com idade média de 32 anos.5
Mutações hereditárias e adquiridas que predispõem à hipercoagubilidade já foram identificadas. Os genes mais frequentemente envolvidos são o MTHFR (methylenetetrahydrofolate reductase), PAI‐1 (plasminogen activator inhibitor‐1), gene da protrombina e do fator V.8 Uma revisão sistemática encontrou a mutação PAI‐1 ‐ 675 4G/5G como a mais frequente (85,26%), enquanto a PAI‐1 A844G, MTHFR C677T e MTHFR A1298C se seguiram em ordem de prevalência, respectivamente. Nos pacientes da Europa, América do Norte e América do Sul, as mutações G20210A no gene da protrombina e G1691A no gene do fator V também foram encontradas em parcelas significantes dos pacientes.8
Condições protrombóticas sistêmicas são consideradas os principais fatores de risco, como a síndrome do anticorpo antifosfolípide (SAAF), as colagenoses (lúpus sistêmicos, artrite reumatoide, síndrome de Sjögren, doença mista do tecido conjuntivo) e a anemia falciforme.9 As comorbidades estatisticamente mais associadas são diabetes, hipertensão arterial sistêmica e doença venosa periférica.10 Na tabela 1 estão destacadas as principais comorbidades associadas à VL.
Comorbidades associadas à vasculopatia livedoide
| Comorbidades | |
| Metabólicas | Diabetes mellitus, hipertrigliceridemia familiar, hipertireoidismo, obesidade, hiper‐homocisteinemia |
| Cardiocirculatórias | Hipertensão arterial sistêmica, insuficiência venosa periférica, doença arterial coronariana |
| Reumatológicas | Artrite reumatoide, lúpus eritematoso sistêmico, síndrome do anticorpo antifosfolípide, doença mista do tecido conjuntivo |
| Hematológicas | Linfomas sistêmicos, gamopatias monoclonais, policitemia vera, trombofilias adquiridas (crioglobiulinas, crioaglutininas, criofibrinogênio), trombofilias congênitas (associadas a mutações nos fatores V, VIII, IX, gene da protrombina, proteínas C e S, antitrombina III, inibidor do fator do plasminogênio e do gene da metilenotetra‐hidrofolato redutase [MTHFR]) |
| Infecciosas | HIV, hepatites B e C |
| Miscelânea | Câncer de mama, asma, migrânea, transplante renal, esclerodermia |
Embora o processo fisiopatológico ainda não esteja totalmente elucidado, a trombose não inflamatória dos vasos da derme parece ser um evento chave. Redução da velocidade do fluxo sanguíneo, dano endotelial e alterações dos fatores de coagulação favorecem a hipercoagulabilidade e atuam de maneira sinérgica para o surgimento da doença. O processo trombótico pode se estender para o vasa nervorum e ocasionar neuropatia.7 A nível molecular, o aumento de fatores protrombóticos, como fatores VIII, IX, anticorpos antifosfolípides, homocisteína e lipoproteína (a) são as alterações mais frequentemente relatados.6,10,11 Criado et al., em estudo com 75 pacientes brasileiros com VL, encontraram que cerca de 66% dos casos apresentavam fatores trombofílicos, em que a lipoproteína(a) foi o fator mais comum detectado.10
As lesões cutâneas ocorrem por deposição pericapilar de fibrina e formação de trombos que atuam como barreira à difusão de oxigênio aos tecidos, causando isquemia e baixa perfusão tecidual, que prejudica a cicatrização. Além disso, a hipóxia torna o local afetado uma barreira ineficaz às bactérias, aumentando o risco de infecção.12
Manifestações clínicasA manifestação clássica inclui pequenas úlceras profundas, necróticas e dolorosas que atingem a região distal das pernas, especialmente a região maleolar e o dorso do pé (fig. 1A). As úlceras cicatrizam lentamente e deixam cicatrizes características branco‐marfínicas atróficas conhecidas como atrofia branca de Millian (fig. 1B). Nesses pacientes, o livedo racemoso pode estar presente, completando uma tríade clássica (fig. 1A).6,7 O livedo reticular também pode ser encontrado, tanto nos membros inferiores quanto, mais raramente, em outras partes do corpo, como mãos, quadril e abdome.4
As úlceras podem ser precedidas por máculas e pápulas purpúricas. Nesse estágio precoce, a dor pode já ser evidente, caracteristicamente intensa e debilitante.4 O quadro álgico pode ser agravado pela presença da neuropatia (em mais de 10% dos pacientes), que deve ser ativamente investigada, particularmente nos casos de dor refratária. O padrão de mononeurite múltipla é o mais prevalente à eletroneuromiografia, determinando sensações dolorosas assimétricas e que não se restringem às áreas ulceradas.13 Embora ocorra com menor frequência, prurido também pode estar presente.4
Na história natural da VL, as lesões recidivam e remitem sem gatilho claro, e é comum que o paciente apresente múltiplas lesões em diferentes estágios de cicatrização.14
Como comentado anteriormente, a VL pode estar associada a estados de hipercoagulabilidade, doenças autoimunes e estase venosa; entretanto, mais da metade dos casos ocorre em indivíduos sem comorbidades.14 É recomendado que, além do exame físico completo, seja solicitada avaliação laboratorial para investigação de possíveis causas sistêmicas associadas – trombofilias, anticorpos antifosfolípides, presença de paraproteínas, rastreio para colagenoses e avaliação das funções hepática e renal (tabela 1).15
Há proposta recente de classificação da VL de acordo com alterações clínicas e laboratoriais presentes: 1) idiopática, sem alterações clínicas e laboratoriais subjacentes; 2) associada a estados de hipercoagulabilidade; 3) associada a doença reumatológica; 4) com doença venosa concomitante; 5) associada a outros mecanismos ou mecanismos multifatoriais.14
HistopatologiaO exame histopatológico é útil para confirmação diagnóstica. Em alguns casos, mais de uma biopsia pode ser necessária.10,15 Os achados clássicos são trombos nos vasos da derme e infiltrado inflamatório linfocítico perivascular esparso. Deposição de material fibrinoide nas paredes dos vasos e na derme também pode ser encontrada (fig. 2).15
.")
A presença de infiltrado neutrofílico não é usual, mas pode estar presente nas biopsias realizadas em áreas ulceradas antigas.10 A identificação de leucocitoclasia aponta para diagnósticos diferenciais que cursam com vasculite.15
A imunofluorescência direta é positiva na maioria dos casos.10,16 A deposição ocorre majoritariamente nos vasos sanguíneos, mas também pode ser visualizada na junção dermoepidérmica. O depósito de IgM e C3 é mais frequente, mas IgG e IgA também podem ocorrer.15 A coleta de material deve ser realizada nas lesões mais recentes, preferencialmente não ulceradas.
Diagnóstico diferencialO diagnóstico diferencial inclui doenças inflamatórias e vaso‐oclusivas que cursam com ulcerações nos membros inferiores. Dentre as doenças inflamatórias, destaca‐se o pioderma gangrenoso e vasculites de médio calibre (poliarterite nodosa ‐ PAN e vasculites associadas ao ANCA).15 Dentre os distúrbios vaso‐oclusivos, calcifilaxia, úlcera hipertensiva de Martorell, endocardite, fenômeno de Lúcio, embolia por colesterol, crioglobulinemia e úlceras induzidas por cocaína contaminada por levamizol devem ser descartados.15
Exame anatomopatológico associado às manifestações clínicas e investigação laboratorial complementar são necessários para a definição diagnóstica. Nos casos em que há neuropatia, o diagnóstico diferencial com PAN deve estar bem estabelecido; para tanto, as biopsias devem atingir as porções mais profundas do subcutâneo para representar os vasos de médio calibre.13
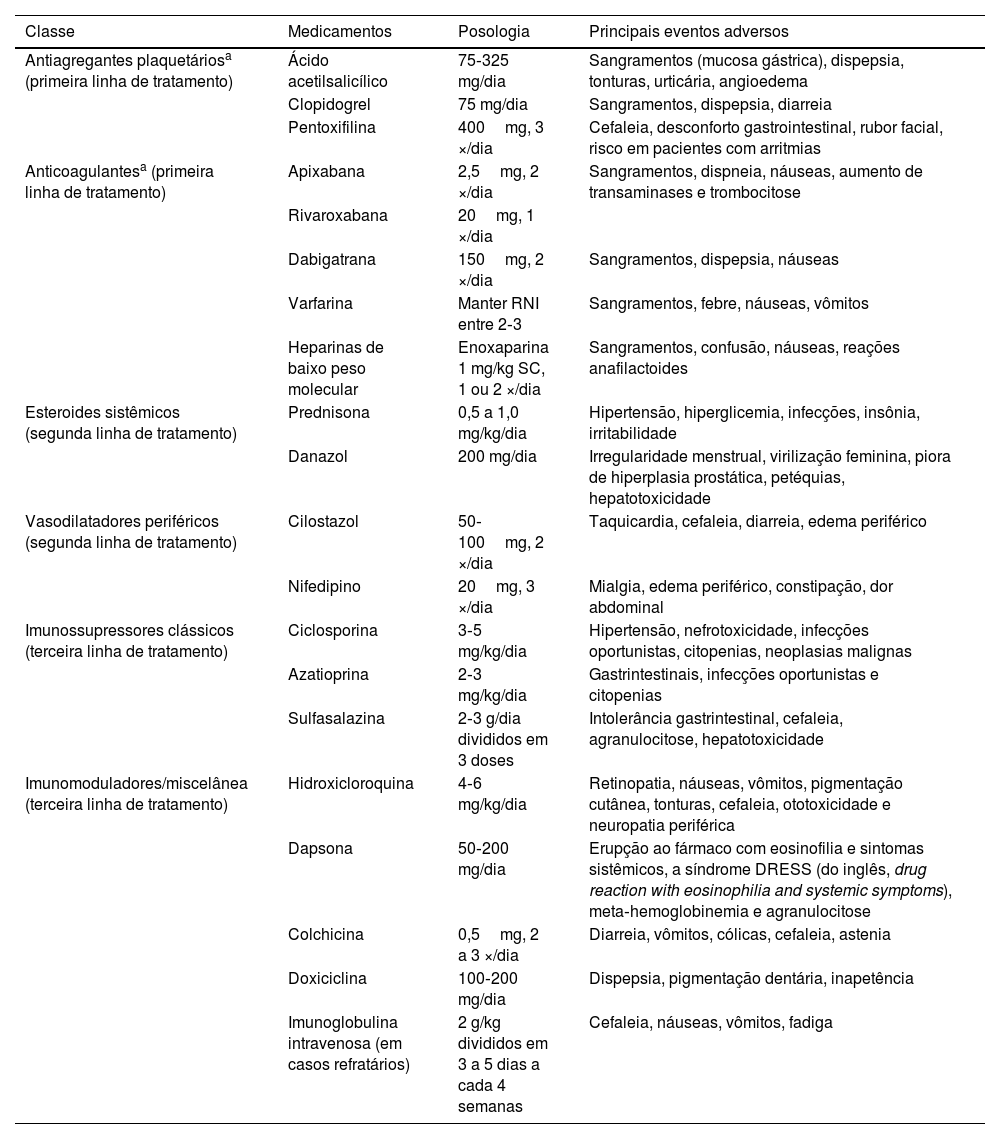
TratamentoO tratamento da VL é desafiador e geralmente requer uma combinação de opções terapêuticas com mecanismos de ação distintos e sinérgicos (tabela 2). Segundo revisão sistemática, a maioria dos estudos classifica a resposta terapêutica como parcial (quando há qualquer melhora que não seja completa), remissão (quando há cicatrização de todas as lesões) e recidiva (quando há novo surto). Segundo essa revisão sistemática, cerca de 44% dos pacientes não apresentam resposta ou não toleraram as primeiras opções terapêuticas.5
Principais tratamentos medicamentosos da vasculopatia livedoide
| Classe | Medicamentos | Posologia | Principais eventos adversos |
|---|---|---|---|
| Antiagregantes plaquetáriosa (primeira linha de tratamento) | Ácido acetilsalicílico | 75‐325 mg/dia | Sangramentos (mucosa gástrica), dispepsia, tonturas, urticária, angioedema |
| Clopidogrel | 75 mg/dia | Sangramentos, dispepsia, diarreia | |
| Pentoxifilina | 400mg, 3 ×/dia | Cefaleia, desconforto gastrointestinal, rubor facial, risco em pacientes com arritmias | |
| Anticoagulantesa (primeira linha de tratamento) | Apixabana | 2,5mg, 2 ×/dia | Sangramentos, dispneia, náuseas, aumento de transaminases e trombocitose |
| Rivaroxabana | 20mg, 1 ×/dia | ||
| Dabigatrana | 150mg, 2 ×/dia | Sangramentos, dispepsia, náuseas | |
| Varfarina | Manter RNI entre 2‐3 | Sangramentos, febre, náuseas, vômitos | |
| Heparinas de baixo peso molecular | Enoxaparina 1 mg/kg SC, 1 ou 2 ×/dia | Sangramentos, confusão, náuseas, reações anafilactoides | |
| Esteroides sistêmicos (segunda linha de tratamento) | Prednisona | 0,5 a 1,0 mg/kg/dia | Hipertensão, hiperglicemia, infecções, insônia, irritabilidade |
| Danazol | 200 mg/dia | Irregularidade menstrual, virilização feminina, piora de hiperplasia prostática, petéquias, hepatotoxicidade | |
| Vasodilatadores periféricos (segunda linha de tratamento) | Cilostazol | 50‐100mg, 2 ×/dia | Taquicardia, cefaleia, diarreia, edema periférico |
| Nifedipino | 20mg, 3 ×/dia | Mialgia, edema periférico, constipação, dor abdominal | |
| Imunossupressores clássicos (terceira linha de tratamento) | Ciclosporina | 3‐5 mg/kg/dia | Hipertensão, nefrotoxicidade, infecções oportunistas, citopenias, neoplasias malignas |
| Azatioprina | 2‐3 mg/kg/dia | Gastrintestinais, infecções oportunistas e citopenias | |
| Sulfasalazina | 2‐3 g/dia divididos em 3 doses | Intolerância gastrintestinal, cefaleia, agranulocitose, hepatotoxicidade | |
| Imunomoduladores/miscelânea (terceira linha de tratamento) | Hidroxicloroquina | 4‐6 mg/kg/dia | Retinopatia, náuseas, vômitos, pigmentação cutânea, tonturas, cefaleia, ototoxicidade e neuropatia periférica |
| Dapsona | 50‐200 mg/dia | Erupção ao fármaco com eosinofilia e sintomas sistêmicos, a síndrome DRESS (do inglês, drug reaction with eosinophilia and systemic symptoms), meta‐hemoglobinemia e agranulocitose | |
| Colchicina | 0,5mg, 2 a 3 ×/dia | Diarreia, vômitos, cólicas, cefaleia, astenia | |
| Doxiciclina | 100‐200 mg/dia | Dispepsia, pigmentação dentária, inapetência | |
| Imunoglobulina intravenosa (em casos refratários) | 2 g/kg divididos em 3 a 5 dias a cada 4 semanas | Cefaleia, náuseas, vômitos, fadiga |
O tratamento não medicamentoso envolve medidas direcionadas à estase venosa e cessação do tabagismo.7 As terapias compressivas devem ser prescritas assim que a doença arterial periférica seja excluída, pela palpação dos pulsos periféricos, medida do índice tornozelo‐braquial ou exames de imagem, como o Doppler arterial de membros inferiores.15 As comorbidades, como as cardiopatias e o diabetes, devem ter controle otimizado.6,17 Pacientes com hiper‐homocisteinemia podem ser tratados com reposição de ácido fólico e vitaminas B6 e B12.7
Os antiagregantes plaquetários são considerados o primeiro degrau na escalada do tratamento, atuando na prevenção da formação de trombos pela vasodilatação e inibição da ciclo‐oxigenase.5 O ácido acetil salicílico (AAS) deve ser usado na dose de 75 a 325mg/dia.7 O clopidogrel na dose de 75mg/dia também é opção viável.18 A pentoxifilina é uma xantina com propriedades antiagregantes plaquetárias e vasodilatadoras, além de aumentar a flexibilidade dos leucócitos e eritrócitos, permitindo melhor fluxo, usada isoladamente ou em combinação com os demais antiagregantes plaquetários. Cardiopatias graves e hipotensão são contraindicações. A dose usual de pentoxifilina é de 400mg, duas a três vezes ao dia.7 Os efeitos colaterais gastrintestinais podem limitar o uso da pentoxifilina e AAS.10
Os anticoagulantes são os fármacos com melhor resposta em monoterapia, por isso podem ser considerados o tratamento de primeira linha, juntamente com os antiagregantes plaquetários.5,17 Os novos anticoagulantes orais inibidores seletivos do fator de coagulação Xa (apixabana, edoxabana e rivaroxabana) ou da protrombina (dabigatrana) são ótima opção terapêutica. Doses menores do que as usuais podem ser eficazes.19 Essa classe apresenta início rápido de ação e não requer dosagens laboratoriais seriadas.7 O tratamento pode ser contínuo ou intermitente, nos casos menos graves.20 Apesar da praticidade e resolutividade dos novos anticoagulantes orais, a varfarina e as heparinas de baixo peso molecular ainda são opção terapêutica para os pacientes com atividade refratária.6
Esteroides sistêmicos, como prednisolona e danazol, são opção terapêutica adjuvante e podem ser utilizados em associação com os antiagregantes plaquetários e anticoagulantes. Eles apresentam maior eficácia quando os pacientes têm comorbidades reumatológicas, como artrite reumatoide e lúpus eritematoso sistêmico. Além dos efeitos anti‐inflamatórios, os esteroides atuariam acelerando a fibrinólise e inibindo a coagulação.5 O danazol acelera a produção hepática das proteínas C e S, e é especialmente útil em pacientes com aumento de lipoproteína (a). Em geral, é bem tolerado, mas até 20% dos pacientes podem apresentar algum evento adverso, como acentuação de hiperplasia prostática, irregularidade menstrual, virilização de pacientes do sexo feminino e alterações hepáticas, que devem ser monitoradas.5,10
Imunossupressores clássicos e fármacos com ação anti‐inflamatória, como hidroxicloroquina, colchicina, dapsona, doxiciclina, azatioprina, ciclosporina e sulfazalassina, podem ser associados ao tratamento. Usualmente, são prescritos em conjunto com antiagregantes plaquetários e/ou anticoagulantes.5,15
Fármacos vasodilatadores periféricos, como nifedipino e cilostazol, podem melhorar a dor e ajudar a cicatrização.7 Ainda nessa linha de ação, visando a inibição da vasoconstrição simpática, recentemente foi relatada a aplicação de toxina botulínica A perilesional com bons resultados, porém com recidiva das lesões em cerca de três meses.21
Diante do número de opções terapêuticas e da dificuldade de manejo, um grupo de autores tentou racionalizar o manejo inicial combinado por meio da abordagem CHAP (bloqueadores de canais de cálcio, hidroxicloroquina, ácido acetilsalicílico e pentoxifilina), com taxa de resposta total superior a 80% no seguimento de dois anos, mas ressalta‐se o pequeno tamanho amostral desse estudo, com apenas 12 pacientes.22
A imunoglobulina intravenosa (IgIV) é modalidade terapêutica de resgaste reservada aos casos refratários aos tratamentos anteriores.5,7 Embora o mecanismo de ação exato não seja bem compreendido, a inibição da produção de anticorpos antifosfolípides, da agregação plaquetária e da disfunção endotelial poderiam explicar a resposta clínica.5 Uma revisão sistemática encontrou taxa de resposta de 95%, com doses de 1‐2,1g/kg, divididas em dois a três dias consecutivos e repetidas a cada quatro semanas. O intervalo de tratamento foi aumentado e a medicação suspensa, de acordo com a experiência do clínico assistente. O primeiro sinal de resposta clínica foi a melhora da dor, e as ulcerações demoraram um período de dias a semanas para cicatrizar. Recorrência ocorreu em cerca de 1/3 dos pacientes.23 Casos que não respondem a IgIV podem ser tratados com fator ativador de plasminogênio tecidual, mas seu uso requer ambiente hospitalar e estrito controle de complicações hemorrágicas.7
Oxigenoterapia hiperbárica é uma modalidade adjuvante, limitada por disponibilidade e custos, realizada com base na dissolução plasmática de oxigênio promovida pela oferta de oxigênio a 100% sob altas pressões (duas a três vezes a pressão atmosférica), promovendo oxigenação de tecidos hipóxicos.5
O uso de imunobiológicos anti‐TNFα e inibidores da JAK tem sido relatado recentemente para casos refratários, rapidamente progressivos ou com contraindicações ao uso de antiagregantes plaquetários e anticoagulantes. Entretanto, esse uso ainda é incipiente e mais estudos são necessários.15,23–26
Assim, o tratamento de VL ainda é um desafio com várias opções terapêuticas e com taxas de cicatrização variável. Além disso, a maioria dos tratamentos baseia‐se em estudos principalmente tipo relatos de casos e séries de casos, os quais fornecem baixos níveis de evidência.5
CalcifilaxiaIntroduçãoA calcifilaxia, também conhecida como arteriolopatia calcificante urêmica, é vasculopatia rara, cutâneo‐sistêmica e potencialmente fatal. Caracteriza‐se por intensa deposição de cálcio em pequenos vasos sanguíneos da derme profunda e tecido subcutâneo, em geral associada ao hiperparatireoidismo secundário com alteração do metabolismo do cálcio e fósforo. Pacientes com doença renal em estágio terminal (baixa taxa de filtração glomerular ou terapia renal substitutiva) representam a principal população afetada pela doença (calcifilaxia urêmica). Contudo, a presença de doença renal não é requisito absoluto para o diagnóstico, podendo ser observada mesmo na ausência da comorbidade, referida nesses casos como calcifilaxia não urêmica. As úlceras cutâneas intensamente dolorosas e necróticas contribuem com a significante taxa de mortalidade da doença (60%‐80%), ainda que diagnosticada em estágios iniciais.27,28
O termo calcifilaxia surgiu após o professor Hans Selye e seus colaboradores conduzirem, em 1961, experimentos sobre o mecanismo de calcificação metastática. Eles expuseram ratos a doses muito elevadas de vitaminas D ou hormônio da paratireoide, associado a dieta rica em fósforo ou a administração intravenosa/intraperitoneal de sais de ferro. Desse modo, foram capazes de demonstrar calcificação metastática maciça do tecido subcutâneo. Nesse experimento, o desenvolvimento de calcificação cutânea foi considerado reação adaptativa ou filática e foi referido como calcifilaxia (calcificação e filaxia).29,30 Entretanto, nesse modelo experimental não foi demonstrada aterosclerose com calcificação medial, atualmente considerada a fisiopatologia básica da calcifilaxia. Até os dias atuais, o termo calcifilaxia é utilizado amplamente com base nesse experimento, mas é inadequado para explicar a fisiopatogenia da doença.30–32
Após esse experimento, foram publicados relatos de casos que descreviam pacientes com insuficiência renal que desenvolveram calcificações subcutâneas generalizadas. Os autores fizeram correlação desses casos clínicos com o modelo experimental de Selye, diagnosticando esses pacientes com calcifilaxia. Diversos relatos semelhantes foram realizados posteriormente utilizando‐se o termo calcifilaxia, o qual ainda hoje é amplamente utilizado.30
EpidemiologiaA incidência da calcifilaxia em pacientes com terapia renal substitutiva varia de 0,04% a 4%, com aumento crescente na última década. O intervalo entre o início da diálise e o surgimento da doença é de aproximadamente 30 meses. Pacientes tratados com diálise peritoneal apresentam maior incidência em relação aos tratados com hemodiálise. A idade média de diagnóstico varia entre 50‐70 anos, e é rara em crianças. Aproximadamente 60%‐70% dos pacientes com calcifilaxia são do sexo feminino, e alguns estudos relatam maior incidência em caucasianos.33,35
Fisiopatologia e fatores de riscoEmbora a patogênese da calcifilaxia envolva múltiplos fatores etiológicos, a complexa cadeia de eventos resulta da inflamação e do estado de hipercoagulabilidade secundário às alterações do metabolismo de cálcio e fósforo, com consequente calcificação vascular e necrose isquêmica cutânea (fluxograma – fig. 3). O evento inicial é o estreitamento progressivo dos lúmens da microcirculação cutânea e subcutânea por calcificação dentro da camada média (conhecida como calcificação medial), proliferação de células endoteliais e fibrose abaixo da íntima (conhecida como fibroplasia subintimal). Posteriormente, trombose se desenvolve no lúmen do vaso, promovendo a oclusão vascular e as lesões isquêmicas.35

Fluxograma da sequência de eventos relacionados à fisiopatologia da calcifilaxia: calcificação de arteríolas, lesão endotelial e consequente oclusão trombótica desses vasos. Essas alterações causam isquemia e necrose do tecido subcutâneo, com surgimento de úlceras necróticas nos estágios mais avançados. PTH, paratormônio.
A calcificação vascular está associada à falta de inibidores de calcificação molecular na parede do vaso. Um dos principais controladores desse mecanismo é a vitamina K, a qual ativa um potente inibidor de calcificação chamado proteína da matriz Gla (MGP), secretada por células musculares lisas vasculares e células endoteliais.36 Portanto, estados que levam à deficiência de vitamina K favorecem a calcificação vascular. Em pacientes com doença renal em estágio terminal, a deficiência de vitamina K pode ocorrer por menor ingestão de alimentos ricos na substância e também por utilização frequente de anticoagulantes inibidores da vitamina K, como a varfarina.37 Segundo estudo alemão com dados de um grande banco de registro nacional, cerca de 50% dos pacientes com diagnóstico de calcifilaxia e doença renal estavam em uso de varfarina no momento do diagnóstico.36
Pacientes com calcifilaxia apresentam associadamente alta prevalência de hipercoagulabilidade congênita ou adquirida. Essa condição favorece a trombose na microcirculação, que é o evento final que ocasiona a oclusão vascular. As alterações mais comuns são presença do anticorpo anticoagulante lúpico, deficiência de antitrombina e deficiência de proteínas C e S.38
Diversos outros fatores de risco foram identificados, como sexo feminino, etnia caucasiana, diabetes mellitus, obesidade, tempo prolongado de diálise, hipoalbuminemia e alterações do metabolismo do cálcio e fósforo.27,29
Manifestações clínicas e diagnósticoA calcifilaxia pode ser classificada clinicamente como urêmica quando ocorre em pacientes com doença renal crônica terminal, ou não urêmica quando ocorre em pacientes com função renal normal ou que estejam em estágios iniciais de doença renal crônica com fatores de risco como obesidade mórbida, hipertensão arterial e diabetes mellitus tipo 2 como resultado de síndrome metabólica. A calcifilaxia urêmica é a mais frequente. Entretanto, as manifestações clínicas são similares independente da associação com doença renal crônica terminal.3,34 Calcifilaxia deve ser suspeitada em pacientes com os fatores de risco citados anteriormente e que apresentem inicialmente lesões nodulares ou placas subcutâneas dolorosas, purpúricas/eritematosas, associadas a livedo racemoso, principalmente quando presentes em áreas de maior adiposidade, como tronco e membros inferiores. As lesões iniciais rapidamente progridem para úlceras necróticas intensamente dolorosas, com extensão centrífuga, no clássico formato reticulado (fig. 4). Infecção bacteriana secundária pode estar associada e é uma das principais causas de óbito.27,28,30 Embora as lesões cutâneas sejam os principais sinais clínicos da doença, outros órgãos, como pulmões, músculos esqueléticos, pâncreas, cérebro, olhos e trato digestivo também podem estar envolvidos.30
 Púrpura retiforme e estágio avançado de úlceras necróticas no clássico formato reticulado nos membros inferiores. (B) Úlcera necrótica em maior destaque (paciente da ref. 39).")
A história do paciente deve ser obtida e exame completo deve ser realizado para identificar lesões cutâneas adicionais. Em pacientes que usam varfarina, deve ser realizado diagnóstico diferencial entre calcifilaxia e necrose por varfarina. Outros importantes diagnósticos diferenciais são vasculopatia livedoide, úlcera isquêmica hipertensiva, pioderma gangrenoso, púrpura fulminans e vasculites necrotizantes.34
A suspeita clínica é importante para um diagnóstico precoce, e exames laboratoriais podem ajudar na complementação diagnóstica. Elevações dos níveis de cálcio ou fosfato sérico não são sempre encontrados; no entanto, quando alterados, corroboram o diagnóstico.39 Considera‐se alterado quando o produto cálcio/fosfato for maior que 70mg2/dL2. Entretanto, segundo estudo alemão, cerca de 86% dos pacientes dependentes de diálise com calcifilaxia apresentavam níveis séricos de cálcio normais ou baixos e 40% tinham níveis de fosfato normais ou baixos.36
A confirmação diagnóstica é realizada por exame histopatógico por meio da biopsia cutânea na periferia da lesão, com profundidade suficiente para atingir o tecido celular subcutâneo, evitando as áreas necróticas. Os principais achados histológicos constituem: calcificação de pequenos vasos, hiperplasia da íntima e oclusão trombótica de vasos na derme e no tecido subcutâneo. Infiltrado inflamatório é frequentemente observado (fig. 5). Colorações específicas para evidenciar a presença de cálcio no tecido, como a coloração de Von Kossa, são importantes para complementar o diagnóstico.27,40
Tratamento.")
O tratamento da calcifilaxia é multidisciplinar e se dá com base no controle dos fatores de risco, no controle efetivo da dor, na abordagem local das lesões inclusive com tratamento de possível infecção secundária e no tratamento específicos com medicações para estabilizar a doença (tabela 3).33
Principais tratamentos para calcifilaxia
| Medidas gerais | Detalhamento |
|---|---|
| Controle dos fatores de risco | Interrupção do uso de varfarina, vitamina D e fosfato de cálcio |
| Manutenção do nível sérico do hormônio da paratireoide (PTH) entre 150‐300 ng/mL | |
| Cinacalcet para tratamento de hiperparatireoidismo secundário | |
| Intensificação da diálise | |
| Paratireoidectomia se hiperparatireoidismo secundário refratário | |
| Controle da dor | Analgesia com opioides (morfina, codeína e hidrocodona devem ser evitadas porque tem metabólitos ativos que se acumulam na insuficiência renal e causam depressão respiratória) |
| Considerar avaliação da terapia antálgica | |
| Manejo local da úlcera | Desbridamento cirúrgico |
| Desbridamento enzimático (colagenase ou papaína) ou autolítico (hidrocoloide ou hidrogel) | |
| Curativos | |
| Enxertos cutâneos | |
| Oxigenioterapia hiperbárica | |
| Tratamento de infecção secundária | Antibióticos sistêmicos |
| Tratamento específico | Dose: 25g em 100mL de solução administrada por via intravenosa três vezes por semana durante os últimos 30 a 60 minutos de hemodiálise |
| Tiossulfato de sódio | Principais eventos adversos: náuseas, vômitos, acidose metabólica e sepse |
No controle dos fatores de risco, deve‐se interromper o uso de varfarina (antagonista da vitamina K), vitamina D e fosfato de cálcio. A diálise deve ser intensificada para controle do metabolismo mineral até as faixas‐alvo de cálcio, fósforo e hormônio da paratireoide. Outra estratégia é substituir a vitamina D ativada por cinacalcet para atingir o nível‐alvo do hormônio da paratireoide. Paratireoidectomia é indicada nos casos de hiperparatireoidismo refratário grave.33
O tratamento local das lesões é fundamental para melhora da dor e da evolução cicatricial. Os objetivos são remover o exsudado e o tecido necrótico, além de prevenir infecção. O desbridamento cirúrgico dos tecidos necróticos deve ser realizado sempre que possível, apesar do desafio em virtude da dor intensa associada. Outros métodos de desbridamento também são indicados, como agentes desbridantes enzimáticos (colagenase ou papaína) ou autolíticos (hidrocoloide ou hidrogel), evitando traumas teciduais ou excesso de manipulação. A prescrição de antibióticos sistêmicos deve ser realizada na suspeita de infecção. Após estabilização da ferida e presença do tecido de granulação, devem ser utilizados curativos para manutenção do meio úmido ou enxerto de pele de espessura parcial. Há também estudos relatando benefícios com oxigenioterapia hiperbárica.34,41,42
O tratamento específico da calcifilaxia pode ser feito com tiossulfato de sódio (agente vasodilatador capaz de inibir a calcificação vascular pelos adipócitos) e bisfosfonatos (agentes capazes de inibir o transporte de fósforo e reduzir a formação de cristais de fosfato de cálcio).41
Tiossulfato de sódio é o fármaco mais frequentemente utilizado.43 A dose usual é de 25g em 100mL de solução administrada por via intravenosa três vezes por semana durante os últimos 30 a 60 minutos de hemodiálise. A duração ideal do curso do tratamento não é clara. Em revisão sistemática que incluiu relatos de casos e séries de casos, totalizando 358 pacientes com calcifilaxia e utilização de tiossulfato de sódio, 96,1% dos pacientes estavam em diálise e o tratamento foi eficaz em 70,1% dos pacientes. As doses de tiossulfato de cálcio variaram de 5 a 25g e foi administrado três vezes por semana, em média, até a estabilização das lesões. A administração foi intravenosa (70,3%), intraperitoneal (9,8%), nas sessões de diálise (11,1%) e intravenosa em combinação de comprimidos orais (7,3%). Não houve diferença significante na eficácia do tratamento entre as diferentes formas de administração. Os eventos adversos mais relatados foram náuseas e vômitos (17,2%), seguido de sepse (14,1%) e acidose metabólica (7,8%). Entretanto, essa revisão não conseguiu avaliar a eficácia do tiossulfato de sódio utilizado isoladamente, pois alguns pacientes também utilizaram outras intervenções associadas. Além disso, a falta de um grupo de controle e a natureza retrospectiva dos estudos impedem conclusões definitivas sobre a eficácia da medicação.43
Há relato de utilização de bisfosfonatos para pacientes com calcifilaxia urêmica. Uma série de casos prospectiva com 11 pacientes mostra que houve retardo na progressão da calcifilaxia em todos os pacientes, duas a quatro semanas após o início do tratamento; entretanto, mais estudos controlados são necessários para estabelecer seu benefício.44
Em pacientes com diagnóstico confirmado ou altamente provável, as abordagens terapêuticas devem ser instituídas precocemente em virtude da alta taxa de morbidade e mortalidade da doença.3,27,28
Úlcera isquêmica hipertensiva de MartorellDefiniçãoA úlcera de etiologia hipertensiva, também conhecida como úlcera de Martorell, foi descrita pela primeira vez por Martorell, Hines e Farber, na década de 1940.45 Em 1945, Martorell publicou um relato de caso descrevendo úlceras supramaleolares por arteriolite associada a hipertensão arterial grave em quatro mulheres obesas. Em 1946‐1947, Farber e Hines descreveram as alterações histopatológicas e definiram o termo “úlcera de perna isquêmica hipertensiva”.46
Por definição inicial, a úlcera isquêmica hipertensiva (UIH) ocorre na presença de hipertensão arterial sistêmica geralmente de longa data e grave, na ausência de doença arterial oclusiva periférica ou doença venosa nos membros inferiores, embora mais recentemente tenha‐se descrito associação com essas duas comorbidades.47
A UIH caracteriza‐se por úlcera rapidamente progressiva, extremamente dolorosa e frequentemente resistente aos tratamentos tópicos usuais. A doença não deve ser confundida com úlcera arterial, uma vez que nas úlceras hipertensivas há alterações na microcirculação sem ainda comprometer a macrocirculação, e nas úlceras isquêmicas arteriais há comprometimento da micro e macrocirculação.48
Epidemiologia e fatores de riscoA UIH é causa incomum de úlceras crônicas dos membros inferiores, embora seja uma doença subestimada; é difícil encontrar registros de sua prevalência.32 A UIH constitui 3%–4% das causas das úlceras crônicas dos membros inferiores, com prevalência de 0,5%–1% e incidência de quatro a seis casos por mil habitantes/ano.32
A UIH é mais comum em mulheres, principalmente após os 50 anos, e acomete apenas os membros inferiores. É importante a exclusão dos diagnósticos de doença venosa e arterial oclusiva periférica nos membros inferiores que possam justificar outra etiologia para as úlceras.
Como fatores de risco associados, há a hipertensão arterial em 100% dos casos e o diabetes mellitus tipo 2 em aproximadamente 60% dos pacientes.32,45,48,49
FisiopatologiaA hipertensão arterial é doença sistêmica que acarreta alterações vasculares disseminadas, como macro e microangiopatia.50,51 A hipertensão de longa duração com ou sem diabetes mellitus induz a típica arteriosclerose isquêmica de pequenos vasos subcutâneos. A calcificação e hipertrofia da camada íntima das arteríolas subcutâneas leva ao aumento da resistência vascular, diminuição da perfusão tecidual e isquemia local; é possível considerar a úlcera hipertensiva como lesão de órgão alvo. Ocorre também uma interrupção do reflexo vasodilatador fisiológico nas arteríolas distais da região obstruída, diminuindo ainda mais a perfusão tecidual.48,52 É importante destacar a diferença fisiopatogênica da UIH com a úlcera isquêmica arterial – nesta última ocorre redução da pressão de perfusão cutânea em decorrência da redução do fluxo arterial do membro por comprometimento na macrocirculação além da microcirculação.
Manifestações clínicasA manifestação clínica clássica são úlceras nos terços inferiores das pernas, supramaleolares, nas regiões látero‐posterior (fig. 6A), uni ou bilaterais, podendo acometer a região do tendão de Aquiles (fig. 6B).45,49,52 Aproximadamente metade dos indivíduos afetados apresenta manifestação bilateral no curso da doença.48 Weber et al. realizaram um mapeamento da localização de UIH, encontrando maior frequência no terço medial da perna direita, látero‐posterior seguido de látero‐anterior.47 A região peri‐úlcera pode tornar‐se eritemato‐purpúricas, e podem ocorrer lesões satélites e livedo racemoso.45,48,49 As úlceras são extremamente dolorosas, de maneira desproporcional ao seu tamanho, e não há melhora com a mudança de posição da perna,32,45 como por exemplo elevação ou pendência. Inicialmente, a lesão surge como placa eritematosa, que evolui para coloração violácea e então para a úlcera dolorosa, podendo apresentar aspecto necrótico e contorno bizarro52 (fig. 6A). Segundo publicação recente, que descreve as características clínicas de 69 pacientes com UIH, um novo sinal clínico, “o sinal do batom vermelho” (partes da borda interna da úlcera com tecido avermelhado – fig. 6B), pode auxiliar o diagnóstico clínico associado à borda purpúrica e livedo racemoso.53
 Úlcera com leito necrótico e bordas purpúricas na face lateral da perna, e outra úlcera menor com leito com bom tecido de granulação no maléolo lateral. (B) Extensa úlcera na região posterior e de calcâneo, com leito desvitalizado e borda interna com sinal do batom‐vermelho (seta) e borda externa eritemato‐purpúrica.")
Úlceras isquêmicas hipertensivas. (A) Úlcera com leito necrótico e bordas purpúricas na face lateral da perna, e outra úlcera menor com leito com bom tecido de granulação no maléolo lateral. (B) Extensa úlcera na região posterior e de calcâneo, com leito desvitalizado e borda interna com sinal do batom‐vermelho (seta) e borda externa eritemato‐purpúrica.
Os pulsos costumam ser palpáveis, e na maioria dos casos não há sinais de doença arterial obstrutiva periférica, com índice tornozelo‐braquial (ITB) geralmente normal entre 0,9 e 1,3; no entanto, costumam ser acima de 1,0 em virtude do aumento da resistência vascular causada pela hipertensão arterial sistêmica.45,54 Também se caracterizam por ocorrer em pacientes com ausência de sinais de insuficiência venosa crônica. Entretanto, não se exclui a possibilidade de o paciente apresentar mais de uma dessas condições, e pode haver doença arterial oclusiva periférica e insuficiência venosa crônica concomitante, o que dificulta o diagnóstico.52 Na séria de casos de Karppinen et al., 36,2% dos pacientes com UIH apresentavam doença arterial periférica e 18,8% tinham insuficiência venosa crônica.53
DiagnósticoEntre os diagnósticos diferenciais, encontram‐se úlceras venosas e arteriais (mais comuns que as hipertensivas), pioderma gangrenoso (em virtude das bordas violáceas e leito necrótico)55 e calcifilaxia (clínica e histopatologia semelhante).52 Pioderma gangrenoso é o diagnóstico mais erroneamente realizado nas fases iniciais de UIH. Um estudo relata que cerca de 50% dos pacientes com UIH tiveram o diagnóstico inicial de pioderma gangrenoso.48
O diagnóstico UIH deve ser aventado em pacientes com história de hipertensão arterial grave e pode ser um desafio, considerando que a etiologia hipertensiva é menos comum que a venosa e a arterial. Atualmente, não há critérios diagnósticos estabelecidos, mas comorbidades, dor intensa, quadro clínico típico como características da úlcera e localização e exame histopatológico orientam o diagnóstico. A biopsia para o exame anatomopatológico pode auxiliar na diferenciação; entretanto, necessita ser em fuso na borda da úlcera e “profunda” (incluindo todos os níveis da pele até a subcutâneo e eventualmente fáscia).32,49
No exame histopatológico da UIH podem ser evidenciados acantose, necrose da epiderme e derme, bem como vasos do subcutâneo com estenose, calcificação (calcinose medial de Mönckeberg), hipertrofia da camada média, trombose e hialinose subendotelial e alteração da razão parede/lúmen (fig. 7).52,56 Essas alterações histopatológicas também são encontradas na calcifilaxia,3 e a diferenciação entre UIH e calcifilaxia deve ser feita pela história de hipertensão de longa data e de difícil controle, que favorece úlcera hipertensiva. A história de doença renal crônica grave e a relação do cálcio‐fósforo sérico alterada favorecem o diagnóstico de calcifilaxia.3,45,48,49,52 Em contrapartida, na úlcera venosa há espongiose, edema dérmico, hemossiderófagos, fibrose e ausência de necrose, trombose ou hialinose. Na doença arterial oclusiva periférica há epiderme fina, esclerose dérmica, necrose e trombose, sem acantose ou hialinose. O pioderma gangrenoso, outro importante diagnóstico diferencial, geralmente apresenta infiltrado inflamatório neutrofílico intenso e não há calcificação subcutânea ou da túnica média.52,56 Entretanto, se a biopsia realizada em UIH for muito pequena como as realizadas com punch, corre‐se o risco de visualizar apenas características semelhantes a pioderma gangrenoso, tais como infiltrado neutrofílico, e a probabilidade é alta de perder as características típicas da UIH (arteriolosclerose estenótica subcutânea e calcificação medial).55
Tratamento.")
A abordagem terapêutica deve ser multidisciplinar e consiste em regime combinado, incluindo tratamento anti‐hipertensivo, anticoagulação, controle da dor, tratamento cirúrgico e medidas locais direcionadas à úlcera.
O controle da pressão arterial é sempre indicado; entretanto, não é capaz de reverter as alterações teciduais já estabelecidas e atuar efetivamente na cicatrização. Devem ser evitados ou descontinuados betabloqueadores não seletivos, como propranolol, em virtude da piora da vasoconstrição periférica e da cicatrização. Benefícios são relatados com nifedipino (bloqueadores dos canais de cálcio) e inibidores da enzima conversora de angiotensina (IECA).57,58
Anticoagulantes orais estão indicados, de preferência os não antagonistas de vitamina K,3,32 isso porque os antagonistas da vitamina K, amplamente utilizados na anticoagulação oral, também inibem uma proteína protetora da calcificação dependente da vitamina K.52
A dor é de difícil controle, podendo ser de origem nociceptiva e neuropática. É recomendado, de acordo com a intensidade da dor, o uso de analgésicos comuns e opioides ou anticonvulsivantes como gabapentina ou pregabalina. Entretanto, a primeira linha de tratamento para controle da dor é o desbridamento cirúrgico precoce e a realização de enxertos de pele parcial.54 O enxerto com punch é técnica tradicional e minimamente invasiva, que tem sido associada à redução significante e rápida da dor em úlceras com diferentes causas subjacentes, especialmente nas UIH.59
Dentre os cuidados locais com as úlceras, é fundamental o manejo dos tecidos desvitalizados, a manutenção do ambiente úmido ideal e o controle da carga bacteriana. Desbridamento do tecido necrótico pode ser feito por técnica cirúrgica superficial. O uso de becaplermina, um fator de crescimento derivado de plaquetas humanas e utilizado para úlceras diabéticas, não mostrou melhora na dor ou qualidade de vida em relação ao hidrogel, que favorece o desbridamento autolítico.60 Terapia com pressão negativa pode auxiliar no tratamento.57
A terapia compressiva (25‐30mmHg) pode ser instituída assim que houver controle da dor e desde que não haja doença arterial concomitante, pois acelera a cicatrização.
As comorbidades devem ser avaliadas e tratadas, como diabetes mellitus e tabagismo. Outras indicações de tratamento incluem simpatectomia lombar para controle da dor e da pressão sanguínea, e clorpromazina ou isoxsuprina para melhora do fluxo capilar.45,61
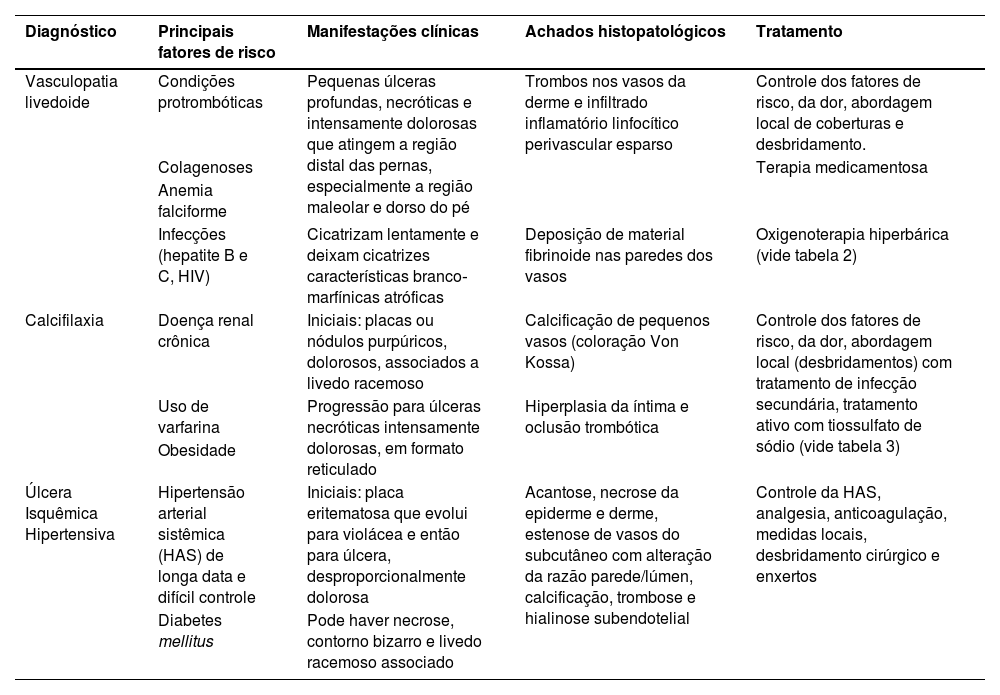
Considerações finaisVL, calcifilaxia e UIH são causas menos comuns de úlceras crônicas, mas seu reconhecimento é fundamental para o correto manejo dos pacientes acometidos. Na tabela 4 encontra‐se um resumo dos principais fatores de risco, manifestação clínica, histopatologia e tratamento relacionados a essas três úlceras isquêmicas por comprometimento da microcirculação dos membros inferiores com a finalidade de resumir o que foi apresentado neste artigo.
Resumo dos principais fatores de risco, manifestação clínica, histopatologia e tratamento de vasculopatia livedoide, calcifilaxia e úlcera isquêmica hipertensiva de Martorell
| Diagnóstico | Principais fatores de risco | Manifestações clínicas | Achados histopatológicos | Tratamento |
|---|---|---|---|---|
| Vasculopatia livedoide | Condições protrombóticas | Pequenas úlceras profundas, necróticas e intensamente dolorosas que atingem a região distal das pernas, especialmente a região maleolar e dorso do pé | Trombos nos vasos da derme e infiltrado inflamatório linfocítico perivascular esparso | Controle dos fatores de risco, da dor, abordagem local de coberturas e desbridamento. |
| Colagenoses | Terapia medicamentosa | |||
| Anemia falciforme | ||||
| Infecções (hepatite B e C, HIV) | Cicatrizam lentamente e deixam cicatrizes características branco‐marfínicas atróficas | Deposição de material fibrinoide nas paredes dos vasos | Oxigenoterapia hiperbárica (vide tabela 2) | |
| Calcifilaxia | Doença renal crônica | Iniciais: placas ou nódulos purpúricos, dolorosos, associados a livedo racemoso | Calcificação de pequenos vasos (coloração Von Kossa) | Controle dos fatores de risco, da dor, abordagem local (desbridamentos) com tratamento de infecção secundária, tratamento ativo com tiossulfato de sódio (vide tabela 3) |
| Uso de varfarina | Progressão para úlceras necróticas intensamente dolorosas, em formato reticulado | Hiperplasia da íntima e oclusão trombótica | ||
| Obesidade | ||||
| Úlcera Isquêmica Hipertensiva | Hipertensão arterial sistêmica (HAS) de longa data e difícil controle | Iniciais: placa eritematosa que evolui para violácea e então para úlcera, desproporcionalmente dolorosa | Acantose, necrose da epiderme e derme, estenose de vasos do subcutâneo com alteração da razão parede/lúmen, calcificação, trombose e hialinose subendotelial | Controle da HAS, analgesia, anticoagulação, medidas locais, desbridamento cirúrgico e enxertos |
| Diabetes mellitus | Pode haver necrose, contorno bizarro e livedo racemoso associado |
Nenhum.
Contribuição dos autoresPriscila Neri Lacerda: Concepção e planejamento do estudo; levantamento bibliográfico, redação do artigo; revisão crítica de literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Lucas Campos Garcia: Concepção e planejamento do estudo; levantamento bibliográfico, redação do artigo; revisão crítica de literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Izabelle Ferreira da Silva Mazeto: Concepção e planejamento do estudo; levantamento bibliográfico, redação do artigo; revisão crítica de literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Hélio Amante Miot: Interpretação dos exames histopatológicos, revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Luciana Patricia Fernandes Abbade: Concepção e planejamento do estudo; levantamento bibliográfico, redação do artigo; revisão crítica de literatura; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Lacerda PN, Garcia LC, Mazeto IFS, Miot HA, Abbade LPF. Livedoid vasculopathy, calciphylaxis, and hypertensive ischemic ulcer: update on ischemic ulcers due to impaired microcirculation of the lower limbs. An Bras Dermatol. 2025;100. https://doi.org/10.1016/j.abd.2024.09.004.
Trabalho realizado no Departamento de Infectologia, Dermatologia, Diagnóstico por Imagem e Radioterapia, Faculdade Medicina, Universidade Estadual Paulista, Botucatu, SP, Brasil.
